气相和水溶液中聚苯并恶嗪氢键网络结构的分子动力学模拟。
IF 2.7
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
摘要
据报道,聚苯并恶嗪(PBZs)曼尼希桥上的胺官能团对其氢键网络结构起着至关重要的作用。然而,人们尚未在水溶液和原子水平上对它们进行深入研究。本研究应用分子动力学模拟研究了由双酚 A/甲胺(m-PBZ)、双酚 A/苯胺基(a-PBZ)和双酚 A/2-(甲氨基)乙胺(e-PBZ)制备的 PBZ 的氢键相互作用的形成。根据模拟结果,PBZ 的氢键网络结构会干扰水分子,导致 PBZ 结构在水溶液中的压实度降低。m-PBZ和a-PBZ结构的氢键种类包括-OH...N(曼尼希)和-OH...O分子内相互作用。然而,对于 e-PBZ 来说,虽然不存在-OH...O 物种,但 2-(乙氨基)乙胺取代基形成的氢键种类却多于 m-PBZ 和 a-PBZ 的氢键种类。此外,在任何水溶液模拟中都没有检测到 PBZ 与水分子之间的分子间氢键相互作用。本文章由计算机程序翻译,如有差异,请以英文原文为准。
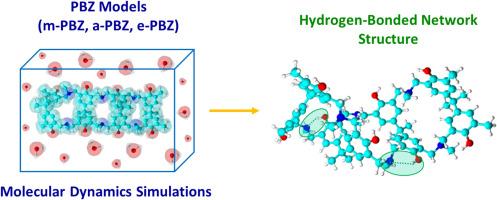
Molecular dynamics simulations of hydrogen-bonded network structures of polybenzoxazines in the gas phase and aqueous solution
The crucial role of the amine functional group at the Mannich bridge of polybenzoxazines (PBZs) has been reported to be responsible for their hydrogen-bonded network structures. However, they have not been thoroughly studied in an aqueous solution and at the atomistic level. In this study, molecular dynamics simulations were applied to investigate the formation of hydrogen bond interactions of PBZs prepared from bisphenol A/methylamine (m-PBZ), bisphenol A/aniline-based (a-PBZ), and bisphenol A/2-(methylamino)ethylamine (e-PBZ). Based on the simulation results, the hydrogen-bonded network structures of the PBZs interfered with water molecules, leading to less compaction of the PBZ structure in the aqueous solution. The hydrogen bonding species of the m-PBZ and a-PBZ structures consisted of the –OH…N (Mannich) and –OH…O intramolecular interactions. However, for e-PBZ, the –OH…O species was not present, but the 2-(ethylamino)ethylamine substituent formed more hydrogen bonding species than those of m-PBZ and a-PBZ. Additionally, the intermolecular hydrogen bond interactions of the PBZs and water molecules were not detected in any of the aqueous solution simulations.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


