{"title":"利用自动方法从同位素追踪数据中量化代谢活动","authors":"Shiyu Liu, Xiaojing Liu, Jason W. Locasale","doi":"10.1038/s42255-024-01144-2","DOIUrl":null,"url":null,"abstract":"<p>Metabolic flux analysis (MFA) is a computational approach to deciphering labelling patterns based on machine learning principles. Differing from typical machine learning algorithms that train a model from known datasets to make predictions, the commonly used MFA algorithm trains a metabolic network with data from isotope tracing experiments and directly outputs the learned information — that is, all fluxes in the network that best fit data<sup>3,5</sup> (Fig. 1b). However, as a machine learning algorithm, current MFA methods often lack systematic evaluation and benchmarking, a standard practice in broader machine learning and artificial intelligence applications<sup>6</sup>. Issues such as algorithmic convergence, flux estimation accuracy and result robustness in MFA studies have been raised but remain largely unaddressed<sup>3</sup>, limiting the effectiveness and broader adoption of these automated tools in metabolic research.</p><p>To advance the capabilities of MFA for complex metabolic networks and extensive isotope tracing datasets, we developed an automated analysis methodology alongside a large-scale metabolic network model. This model comprises over 100 fluxes across key pathways, including glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway (PPP), one-carbon metabolism, and several amino acid (AA) biosynthetic pathways (Fig. 1c, Supplementary Methods). Compared to contemporary MFA tools<sup>7,8,9</sup>, a notable feature of our methodology is the incorporation of organelle compartmentalization, facilitating accurate quantification of exchange fluxes between mitochondria and cytosol in eukaryotic cells (Fig. 1c). While other tools typically require tens of minutes to obtain a solution<sup>7,9</sup>, our methodology can generate an optimized solution, with fluxes that accurately explain the labelling pattern from a <sup>13</sup>C tracing experiment on cultured cell lines, within 2 s on a desktop computer<sup>10</sup> (Supplementary Fig. 1a–d). Nonetheless, a challenge arose from the observation that these optimized solutions could diverge significantly, showing considerable variability in certain net fluxes even with similar loss values (Fig. 1d, Supplementary Fig. 1e,f).To address this problem, we developed an optimization-averaging algorithm that refines the computation process by selecting a subset of solutions with minimal loss (selected solutions) from the pool of optimized solutions and averaging them to produce a new, more stable solution set (averaged solutions) (Fig. 1e, Supplementary Methods). These solutions, along with those generated using the typical strategy used in contemporary software (Supplementary Methods, Supplementary Fig. 1c), were benchmarked using simulated <sup>13</sup>C tracing datasets generated from a known flux vector (Supplementary Fig. 2a). The results demonstrated that, relative to the benchmark, the optimization-averaging algorithm effectively reduced flux variability and improved the accuracy of the results in approximating the known flux, even with varying levels of data availability (Fig. 1f, Supplementary Figs. 2b–e and 3a–e).</p>","PeriodicalId":19038,"journal":{"name":"Nature metabolism","volume":"5 1","pages":""},"PeriodicalIF":18.9000,"publicationDate":"2024-10-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Quantitation of metabolic activity from isotope tracing data using automated methodology\",\"authors\":\"Shiyu Liu, Xiaojing Liu, Jason W. Locasale\",\"doi\":\"10.1038/s42255-024-01144-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Metabolic flux analysis (MFA) is a computational approach to deciphering labelling patterns based on machine learning principles. Differing from typical machine learning algorithms that train a model from known datasets to make predictions, the commonly used MFA algorithm trains a metabolic network with data from isotope tracing experiments and directly outputs the learned information — that is, all fluxes in the network that best fit data<sup>3,5</sup> (Fig. 1b). However, as a machine learning algorithm, current MFA methods often lack systematic evaluation and benchmarking, a standard practice in broader machine learning and artificial intelligence applications<sup>6</sup>. Issues such as algorithmic convergence, flux estimation accuracy and result robustness in MFA studies have been raised but remain largely unaddressed<sup>3</sup>, limiting the effectiveness and broader adoption of these automated tools in metabolic research.</p><p>To advance the capabilities of MFA for complex metabolic networks and extensive isotope tracing datasets, we developed an automated analysis methodology alongside a large-scale metabolic network model. This model comprises over 100 fluxes across key pathways, including glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway (PPP), one-carbon metabolism, and several amino acid (AA) biosynthetic pathways (Fig. 1c, Supplementary Methods). Compared to contemporary MFA tools<sup>7,8,9</sup>, a notable feature of our methodology is the incorporation of organelle compartmentalization, facilitating accurate quantification of exchange fluxes between mitochondria and cytosol in eukaryotic cells (Fig. 1c). While other tools typically require tens of minutes to obtain a solution<sup>7,9</sup>, our methodology can generate an optimized solution, with fluxes that accurately explain the labelling pattern from a <sup>13</sup>C tracing experiment on cultured cell lines, within 2 s on a desktop computer<sup>10</sup> (Supplementary Fig. 1a–d). Nonetheless, a challenge arose from the observation that these optimized solutions could diverge significantly, showing considerable variability in certain net fluxes even with similar loss values (Fig. 1d, Supplementary Fig. 1e,f).To address this problem, we developed an optimization-averaging algorithm that refines the computation process by selecting a subset of solutions with minimal loss (selected solutions) from the pool of optimized solutions and averaging them to produce a new, more stable solution set (averaged solutions) (Fig. 1e, Supplementary Methods). These solutions, along with those generated using the typical strategy used in contemporary software (Supplementary Methods, Supplementary Fig. 1c), were benchmarked using simulated <sup>13</sup>C tracing datasets generated from a known flux vector (Supplementary Fig. 2a). The results demonstrated that, relative to the benchmark, the optimization-averaging algorithm effectively reduced flux variability and improved the accuracy of the results in approximating the known flux, even with varying levels of data availability (Fig. 1f, Supplementary Figs. 2b–e and 3a–e).</p>\",\"PeriodicalId\":19038,\"journal\":{\"name\":\"Nature metabolism\",\"volume\":\"5 1\",\"pages\":\"\"},\"PeriodicalIF\":18.9000,\"publicationDate\":\"2024-10-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature metabolism\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s42255-024-01144-2\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature metabolism","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s42255-024-01144-2","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Quantitation of metabolic activity from isotope tracing data using automated methodology
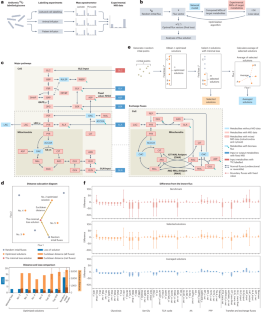
Metabolic flux analysis (MFA) is a computational approach to deciphering labelling patterns based on machine learning principles. Differing from typical machine learning algorithms that train a model from known datasets to make predictions, the commonly used MFA algorithm trains a metabolic network with data from isotope tracing experiments and directly outputs the learned information — that is, all fluxes in the network that best fit data3,5 (Fig. 1b). However, as a machine learning algorithm, current MFA methods often lack systematic evaluation and benchmarking, a standard practice in broader machine learning and artificial intelligence applications6. Issues such as algorithmic convergence, flux estimation accuracy and result robustness in MFA studies have been raised but remain largely unaddressed3, limiting the effectiveness and broader adoption of these automated tools in metabolic research.
To advance the capabilities of MFA for complex metabolic networks and extensive isotope tracing datasets, we developed an automated analysis methodology alongside a large-scale metabolic network model. This model comprises over 100 fluxes across key pathways, including glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway (PPP), one-carbon metabolism, and several amino acid (AA) biosynthetic pathways (Fig. 1c, Supplementary Methods). Compared to contemporary MFA tools7,8,9, a notable feature of our methodology is the incorporation of organelle compartmentalization, facilitating accurate quantification of exchange fluxes between mitochondria and cytosol in eukaryotic cells (Fig. 1c). While other tools typically require tens of minutes to obtain a solution7,9, our methodology can generate an optimized solution, with fluxes that accurately explain the labelling pattern from a 13C tracing experiment on cultured cell lines, within 2 s on a desktop computer10 (Supplementary Fig. 1a–d). Nonetheless, a challenge arose from the observation that these optimized solutions could diverge significantly, showing considerable variability in certain net fluxes even with similar loss values (Fig. 1d, Supplementary Fig. 1e,f).To address this problem, we developed an optimization-averaging algorithm that refines the computation process by selecting a subset of solutions with minimal loss (selected solutions) from the pool of optimized solutions and averaging them to produce a new, more stable solution set (averaged solutions) (Fig. 1e, Supplementary Methods). These solutions, along with those generated using the typical strategy used in contemporary software (Supplementary Methods, Supplementary Fig. 1c), were benchmarked using simulated 13C tracing datasets generated from a known flux vector (Supplementary Fig. 2a). The results demonstrated that, relative to the benchmark, the optimization-averaging algorithm effectively reduced flux variability and improved the accuracy of the results in approximating the known flux, even with varying levels of data availability (Fig. 1f, Supplementary Figs. 2b–e and 3a–e).
期刊介绍:
Nature Metabolism is a peer-reviewed scientific journal that covers a broad range of topics in metabolism research. It aims to advance the understanding of metabolic and homeostatic processes at a cellular and physiological level. The journal publishes research from various fields, including fundamental cell biology, basic biomedical and translational research, and integrative physiology. It focuses on how cellular metabolism affects cellular function, the physiology and homeostasis of organs and tissues, and the regulation of organismal energy homeostasis. It also investigates the molecular pathophysiology of metabolic diseases such as diabetes and obesity, as well as their treatment. Nature Metabolism follows the standards of other Nature-branded journals, with a dedicated team of professional editors, rigorous peer-review process, high standards of copy-editing and production, swift publication, and editorial independence. The journal has a high impact factor, has a certain influence in the international area, and is deeply concerned and cited by the majority of scholars.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


