{"title":"关于 LaCO 和 AcCO 的基态和激发电子态","authors":"Isuru R. Ariyarathna","doi":"10.1039/D4CP03132F","DOIUrl":null,"url":null,"abstract":"<p >High-level <em>ab initio</em> electronic structure analysis of correlated lanthanide- and actinide-based species is laborious to perform and consequently limited in the literature. In the present work, the ground and electronically excited states of LaCO and AcCO molecules were explored utilizing the multireference configuration interaction (MRCI), Davidson corrected MRCI (MRCI+Q), and coupled cluster singles doubles and perturbative triples [CCSD(T)] quantum chemical tools conjoined with correlation consistent triple-ζ and quadruple-ζ quality all-electron Douglas–Kroll (DK) basis sets. The full potential energy curves (PECs), dissociation energies (<em>D</em><small><sub>e</sub></small>s), excitation energies (<em>T</em><small><sub>e</sub></small>s), bond lengths (<em>r</em><small><sub>e</sub></small>s), harmonic vibrational frequencies (<em>ω</em><small><sub>e</sub></small>s), and chemical bonding patterns of low-lying electronic states of LaCO and AcCO are introduced. The ground electronic state of LaCO is a <small><sup>4</sup></small>Σ<small><sup>−</sup></small> (1σ<small><sup>1</sup></small>1π<small><sup>2</sup></small>) which is a product of the reaction between excited La(<small><sup>4</sup></small>F) <em>versus</em> CO(X<small><sup>1</sup></small>Σ<small><sup>+</sup></small>), whereas the ground state of AcCO is a 1<small><sup>2</sup></small>Π (1σ<small><sup>2</sup></small>1π<small><sup>1</sup></small>) deriving from ground state fragments Ac(<small><sup>2</sup></small>D) + CO(X<small><sup>1</sup></small>Σ<small><sup>+</sup></small>). The spin–orbit ground states of LaCO (1<small><sup>4</sup></small>Σ<small><sup>−</sup></small><small><sub>3/2</sub></small>) and AcCO (1<small><sup>2</sup></small>Π<small><sub>1/2</sub></small>) bear ∼13 and 5 kcal mol<small><sup>−1</sup></small><em>D</em><small><sub>0</sub></small> values, respectively. At the MRCI level, the spin–orbit curves, the spin–orbit mixing, and the <em>T</em><small><sub>e</sub></small>s of spin–orbit states of LaCO and AcCO were also analyzed. Lastly, the density functional theory (DFT) calculations were performed applying 16 exchange–correlation functionals that span three rungs of “Jacob's ladder” of density functional approximations (DFAs) to assess DFT errors associated on the <em>D</em><small><sub>e</sub></small> and ionization energy (IE) of LaCO.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 45","pages":" 28337-28348"},"PeriodicalIF":2.9000,"publicationDate":"2024-10-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/cp/d4cp03132f?page=search","citationCount":"0","resultStr":"{\"title\":\"On the ground and excited electronic states of LaCO and AcCO†\",\"authors\":\"Isuru R. Ariyarathna\",\"doi\":\"10.1039/D4CP03132F\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >High-level <em>ab initio</em> electronic structure analysis of correlated lanthanide- and actinide-based species is laborious to perform and consequently limited in the literature. In the present work, the ground and electronically excited states of LaCO and AcCO molecules were explored utilizing the multireference configuration interaction (MRCI), Davidson corrected MRCI (MRCI+Q), and coupled cluster singles doubles and perturbative triples [CCSD(T)] quantum chemical tools conjoined with correlation consistent triple-ζ and quadruple-ζ quality all-electron Douglas–Kroll (DK) basis sets. The full potential energy curves (PECs), dissociation energies (<em>D</em><small><sub>e</sub></small>s), excitation energies (<em>T</em><small><sub>e</sub></small>s), bond lengths (<em>r</em><small><sub>e</sub></small>s), harmonic vibrational frequencies (<em>ω</em><small><sub>e</sub></small>s), and chemical bonding patterns of low-lying electronic states of LaCO and AcCO are introduced. The ground electronic state of LaCO is a <small><sup>4</sup></small>Σ<small><sup>−</sup></small> (1σ<small><sup>1</sup></small>1π<small><sup>2</sup></small>) which is a product of the reaction between excited La(<small><sup>4</sup></small>F) <em>versus</em> CO(X<small><sup>1</sup></small>Σ<small><sup>+</sup></small>), whereas the ground state of AcCO is a 1<small><sup>2</sup></small>Π (1σ<small><sup>2</sup></small>1π<small><sup>1</sup></small>) deriving from ground state fragments Ac(<small><sup>2</sup></small>D) + CO(X<small><sup>1</sup></small>Σ<small><sup>+</sup></small>). The spin–orbit ground states of LaCO (1<small><sup>4</sup></small>Σ<small><sup>−</sup></small><small><sub>3/2</sub></small>) and AcCO (1<small><sup>2</sup></small>Π<small><sub>1/2</sub></small>) bear ∼13 and 5 kcal mol<small><sup>−1</sup></small><em>D</em><small><sub>0</sub></small> values, respectively. At the MRCI level, the spin–orbit curves, the spin–orbit mixing, and the <em>T</em><small><sub>e</sub></small>s of spin–orbit states of LaCO and AcCO were also analyzed. Lastly, the density functional theory (DFT) calculations were performed applying 16 exchange–correlation functionals that span three rungs of “Jacob's ladder” of density functional approximations (DFAs) to assess DFT errors associated on the <em>D</em><small><sub>e</sub></small> and ionization energy (IE) of LaCO.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 45\",\"pages\":\" 28337-28348\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-10-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2024/cp/d4cp03132f?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp03132f\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp03132f","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
On the ground and excited electronic states of LaCO and AcCO†
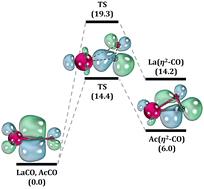
High-level ab initio electronic structure analysis of correlated lanthanide- and actinide-based species is laborious to perform and consequently limited in the literature. In the present work, the ground and electronically excited states of LaCO and AcCO molecules were explored utilizing the multireference configuration interaction (MRCI), Davidson corrected MRCI (MRCI+Q), and coupled cluster singles doubles and perturbative triples [CCSD(T)] quantum chemical tools conjoined with correlation consistent triple-ζ and quadruple-ζ quality all-electron Douglas–Kroll (DK) basis sets. The full potential energy curves (PECs), dissociation energies (Des), excitation energies (Tes), bond lengths (res), harmonic vibrational frequencies (ωes), and chemical bonding patterns of low-lying electronic states of LaCO and AcCO are introduced. The ground electronic state of LaCO is a 4Σ− (1σ11π2) which is a product of the reaction between excited La(4F) versus CO(X1Σ+), whereas the ground state of AcCO is a 12Π (1σ21π1) deriving from ground state fragments Ac(2D) + CO(X1Σ+). The spin–orbit ground states of LaCO (14Σ−3/2) and AcCO (12Π1/2) bear ∼13 and 5 kcal mol−1D0 values, respectively. At the MRCI level, the spin–orbit curves, the spin–orbit mixing, and the Tes of spin–orbit states of LaCO and AcCO were also analyzed. Lastly, the density functional theory (DFT) calculations were performed applying 16 exchange–correlation functionals that span three rungs of “Jacob's ladder” of density functional approximations (DFAs) to assess DFT errors associated on the De and ionization energy (IE) of LaCO.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


