Irene Bertozzi, Andrea Benetti, Elisabetta Cosi, Martina Zerbinati, Cecilia Fortino, Maria Luigia Randi, Paolo Simioni
{"title":"HFE 基因突变对特发性红细胞增多症患者血栓风险的影响:单中心研究","authors":"Irene Bertozzi, Andrea Benetti, Elisabetta Cosi, Martina Zerbinati, Cecilia Fortino, Maria Luigia Randi, Paolo Simioni","doi":"10.1002/jha2.1019","DOIUrl":null,"url":null,"abstract":"<p>Idiopathic erythrocytosis (IE) is characterized by an increase in red blood cell mass without an identified cause. Diagnosis of IE is based on exclusion of all the known forms of primary and secondary acquired erythrocytosis and various congenital primary and secondary polycythaemias [<span>1-3</span>]. Recent studies have demonstrated a genetic complexity of IE, detecting the presence of several genetic variants in genes involved or suspected of being involved in erythrocytosis [<span>4, 5</span>]. In particular, <i>HFE</i> mutations are frequently observed in patients with IE, postulating that iron metabolism impairment is a possible underlying cause for erythrocytosis [<span>6, 7</span>].</p><p>IE shows a peculiar clinical phenotype (male, young, isolated erythrocytosis), a trend for a stable disease with no tendency to spontaneous progression to myelofibrosis or acute leukaemia, but a relevant risk of thrombosis, especially arterial events, also in young patients [<span>5, 8, 9</span>]. To date, no clear factors related to the increased thrombotic risk in IE have been established, therefore current therapeutic indications are aimed only at the management of cardiovascular risk factors. It has been shown that high haematocrit independently promotes arterial thrombosis by increasing the rate of platelet deposition and thrombus growth in spite of the absence of a clonal disease [<span>10</span>], but the role of mutational status in thrombotic risk assessment has never been explored in patients with IE.</p><p>We studied 100 patients referred to our department, with a diagnosis of IE and an available complete medical history, including common cardiovascular risk factors (hypertension, diabetes, dyslipidaemia and active smoking). None of them carried <i>JAK2</i> V617F or exon 12 mutations [<span>1</span>]. Congenital primary and secondary polycythaemias were excluded in the absence of a familial pattern (i.e., at least one relative with erythrocytosis) and known mutations in <i>EPO-R</i> or Oxygen Sensing Pathway genes [<span>2, 3</span>]. A targeted next-generation sequencing (NGS) panel for patients with unexplained erythrocytosis was set up, including genes involved or suspected to be involved in erythrocytosis (Supporting information). Clinical and laboratory data of the patients are shown in Table 1.</p><p>All patients gave written informed consent. The protocol was approved by the local Institutional Ethical Committee (Azienda Ospedaliera di Padova, ref: 3922/AO/16). The study was conducted in compliance with the principles of the Declaration of Helsinki. The statistical tests adopted were logistic regression model for univariate and bivariate analysis and Cox regression model for survival analysis. Survival curve has been prepared with Kaplan–Meier method and compared with log rank test.</p><p>Sixty-seven (67%) patients carry at least one gene variant detected by the NGS study (Table S1). Forty-seven (47%) patients carry at least a mutation of <i>HFE</i> (30 heterozygous and 3 homozygous for H63D variant, 7 heterozygous for C282Y mutation, 2 heterozygous for C65S mutation, 3 compound heterozygous C282Y/H63D, 1 H63D/C65S and 1 C282Y/C65S). No differences in clinical and laboratory parameters have been found comparing patients with or without at least one gene variant detectable in the NGS study. Furthermore, among mutated patients no difference has been observed comparing clinical and laboratory parameters of the different detected variants. We observed 15 vascular events, 11 arterial (6 acute MI, 3 TIA, and 2 ischemic stroke) and 4 venous (2 DVT and 2 PE) thromboses in 13 patients (13%). In nine patients thrombotic event occurs at diagnosis or first evidence of erythrocytosis, in the other four during follow-up after a median time of 4.84 years. Total thrombosis rate in our cohort was 2.51 events*100 pats/years. In univariate analysis, patients with thrombotic complications were older (median age 64 vs. 56 y; <i>p</i> = 0.007, OR 1.071, CI 95% 1.02–1.13), had higher haematocrit (53 vs. 51%; <i>p</i> = 0.027, OR 1.31, CI 95% 1.03–1.66) and higher prevalence of <i>HFE</i> mutations [10 (77%) vs. 34 (39%), <i>p</i> = 0.028, OR 4.61, CI95% 1.18–18.02], while no difference has been reported in gender, haemoglobin and ferritin levels and prevalence of cardiovascular risk factors (Table S2). In bivariate analysis, both the presence of at least one <i>HFE</i> mutation (<i>p</i> = 0.043, OR 4.31, CI 95% 1.05–17.77) and older age at diagnosis (<i>p</i> = 0.011, OR 1.066, CI 95% 1.02–1.12) have been confirmed as risk factors for thrombosis. No difference was demonstrated in the occurrence of thrombotic complications comparing mutated and unmutated patients considering all detected variants in NGS, but stratifying patients on the basis of <i>HFE</i> mutational status (47 patients with at least one <i>HFE</i> mutation vs. 53 <i>HFE</i> wild-type patients) we found a significantly higher frequency of thrombotic complications in patients with at least one <i>HFE</i> mutation (<i>n</i> = 10, 21.3%) compared to all <i>HFE</i> wild-type patients (<i>n</i> = 3, 5.7%; <i>p</i> 0.03). Patients with and without <i>HFE</i> mutation had similar parameters at diagnosis and similar prevalence of cardiovascular risk factors. <i>HFE</i> mutated and wild-type patients showed an incidence of thrombosis of 4.4 and 1.28 events*100, respectively, with a relative risk of 3.44. Patients with <i>HFE</i> mutations showed a worse thrombosis-free survival compared to the wild-type patients (<i>p</i> 0.02; Figure 1), this observation was confirmed also in Cox regression analysis (<i>p</i> = 0.041, HR 5.04, CI 95% 1.07–23.80).</p><p>IE is an indolent disease with a relevant thrombotic risk, lower than polycythaemia vera but higher than the general population [<span>8</span>]. In other forms of erythrocytosis, specific mutations have been shown to affect thrombotic risk assessment [<span>11</span>], but to date the role of mutational status in vascular complications of patients with IE has not yet been evaluated. The impact of <i>HFE</i> mutation on thrombotic risk has been evaluated in other cohorts. <i>HFE</i> mutations, in particular the C282Y mutation, do not seem to worsen both arterial and venous thrombotic risk in patients with hereditary hemochromatosis [<span>12, 13</span>]. Furthermore, it has been reported that the presence of <i>HFE</i> mutations does not impact thrombotic risk in patients with hereditary thrombophilia related to the presence of factor V Leiden [<span>14, 15</span>].</p><p>Although high haematocrit is certainly one of the promoters of increased thrombotic risk in patients with IE compared to the general population [<span>8, 10</span>], our study suggests that the presence of <i>HFE</i> mutations may be an additional risk factor for thrombotic complications, which in our series leads to a more than four-fold increase in thrombotic risk. The mechanism behind this finding is unclear. Considering that patients with hereditary hemochromatosis do not appear to have an increased thrombotic risk [<span>12-15</span>], the combined effect of dysregulation of iron metabolism due to <i>HFE</i> mutation and increased haematocrit may play a role. To date, it seems premature to recommend, in daily clinical practice, the inclusion of screening for <i>HFE</i> mutations in the diagnostic work-up of patients with IE. However, our observations, if confirmed on larger series, may improve risk stratification in order to achieve a more tailored diagnostic and therapeutic approach in patients with IE.</p><p>Irene Bertozzi and Andrea Benetti conceived the study, collected clinical data and wrote the paper. Elisabetta Cosi, Cecilia Fortino and Martina Zerbinati collected clinical data. Maria Luigia Randi and Paolo Simioni conceived the study, supervised research activities and wrote the paper.</p><p>The authors declare no conflicts of interest.</p><p>The authors received no specific funding for this work.</p><p>Informed consent has been obtained from patients included in the study.</p>","PeriodicalId":72883,"journal":{"name":"EJHaem","volume":"5 5","pages":"1086-1088"},"PeriodicalIF":0.0000,"publicationDate":"2024-10-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jha2.1019","citationCount":"0","resultStr":"{\"title\":\"Impact of HFE mutations on thrombotic risk in patients with idiopathic erythrocytosis: A single-centre study\",\"authors\":\"Irene Bertozzi, Andrea Benetti, Elisabetta Cosi, Martina Zerbinati, Cecilia Fortino, Maria Luigia Randi, Paolo Simioni\",\"doi\":\"10.1002/jha2.1019\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Idiopathic erythrocytosis (IE) is characterized by an increase in red blood cell mass without an identified cause. Diagnosis of IE is based on exclusion of all the known forms of primary and secondary acquired erythrocytosis and various congenital primary and secondary polycythaemias [<span>1-3</span>]. Recent studies have demonstrated a genetic complexity of IE, detecting the presence of several genetic variants in genes involved or suspected of being involved in erythrocytosis [<span>4, 5</span>]. In particular, <i>HFE</i> mutations are frequently observed in patients with IE, postulating that iron metabolism impairment is a possible underlying cause for erythrocytosis [<span>6, 7</span>].</p><p>IE shows a peculiar clinical phenotype (male, young, isolated erythrocytosis), a trend for a stable disease with no tendency to spontaneous progression to myelofibrosis or acute leukaemia, but a relevant risk of thrombosis, especially arterial events, also in young patients [<span>5, 8, 9</span>]. To date, no clear factors related to the increased thrombotic risk in IE have been established, therefore current therapeutic indications are aimed only at the management of cardiovascular risk factors. It has been shown that high haematocrit independently promotes arterial thrombosis by increasing the rate of platelet deposition and thrombus growth in spite of the absence of a clonal disease [<span>10</span>], but the role of mutational status in thrombotic risk assessment has never been explored in patients with IE.</p><p>We studied 100 patients referred to our department, with a diagnosis of IE and an available complete medical history, including common cardiovascular risk factors (hypertension, diabetes, dyslipidaemia and active smoking). None of them carried <i>JAK2</i> V617F or exon 12 mutations [<span>1</span>]. Congenital primary and secondary polycythaemias were excluded in the absence of a familial pattern (i.e., at least one relative with erythrocytosis) and known mutations in <i>EPO-R</i> or Oxygen Sensing Pathway genes [<span>2, 3</span>]. A targeted next-generation sequencing (NGS) panel for patients with unexplained erythrocytosis was set up, including genes involved or suspected to be involved in erythrocytosis (Supporting information). Clinical and laboratory data of the patients are shown in Table 1.</p><p>All patients gave written informed consent. The protocol was approved by the local Institutional Ethical Committee (Azienda Ospedaliera di Padova, ref: 3922/AO/16). The study was conducted in compliance with the principles of the Declaration of Helsinki. The statistical tests adopted were logistic regression model for univariate and bivariate analysis and Cox regression model for survival analysis. Survival curve has been prepared with Kaplan–Meier method and compared with log rank test.</p><p>Sixty-seven (67%) patients carry at least one gene variant detected by the NGS study (Table S1). Forty-seven (47%) patients carry at least a mutation of <i>HFE</i> (30 heterozygous and 3 homozygous for H63D variant, 7 heterozygous for C282Y mutation, 2 heterozygous for C65S mutation, 3 compound heterozygous C282Y/H63D, 1 H63D/C65S and 1 C282Y/C65S). No differences in clinical and laboratory parameters have been found comparing patients with or without at least one gene variant detectable in the NGS study. Furthermore, among mutated patients no difference has been observed comparing clinical and laboratory parameters of the different detected variants. We observed 15 vascular events, 11 arterial (6 acute MI, 3 TIA, and 2 ischemic stroke) and 4 venous (2 DVT and 2 PE) thromboses in 13 patients (13%). In nine patients thrombotic event occurs at diagnosis or first evidence of erythrocytosis, in the other four during follow-up after a median time of 4.84 years. Total thrombosis rate in our cohort was 2.51 events*100 pats/years. In univariate analysis, patients with thrombotic complications were older (median age 64 vs. 56 y; <i>p</i> = 0.007, OR 1.071, CI 95% 1.02–1.13), had higher haematocrit (53 vs. 51%; <i>p</i> = 0.027, OR 1.31, CI 95% 1.03–1.66) and higher prevalence of <i>HFE</i> mutations [10 (77%) vs. 34 (39%), <i>p</i> = 0.028, OR 4.61, CI95% 1.18–18.02], while no difference has been reported in gender, haemoglobin and ferritin levels and prevalence of cardiovascular risk factors (Table S2). In bivariate analysis, both the presence of at least one <i>HFE</i> mutation (<i>p</i> = 0.043, OR 4.31, CI 95% 1.05–17.77) and older age at diagnosis (<i>p</i> = 0.011, OR 1.066, CI 95% 1.02–1.12) have been confirmed as risk factors for thrombosis. No difference was demonstrated in the occurrence of thrombotic complications comparing mutated and unmutated patients considering all detected variants in NGS, but stratifying patients on the basis of <i>HFE</i> mutational status (47 patients with at least one <i>HFE</i> mutation vs. 53 <i>HFE</i> wild-type patients) we found a significantly higher frequency of thrombotic complications in patients with at least one <i>HFE</i> mutation (<i>n</i> = 10, 21.3%) compared to all <i>HFE</i> wild-type patients (<i>n</i> = 3, 5.7%; <i>p</i> 0.03). Patients with and without <i>HFE</i> mutation had similar parameters at diagnosis and similar prevalence of cardiovascular risk factors. <i>HFE</i> mutated and wild-type patients showed an incidence of thrombosis of 4.4 and 1.28 events*100, respectively, with a relative risk of 3.44. Patients with <i>HFE</i> mutations showed a worse thrombosis-free survival compared to the wild-type patients (<i>p</i> 0.02; Figure 1), this observation was confirmed also in Cox regression analysis (<i>p</i> = 0.041, HR 5.04, CI 95% 1.07–23.80).</p><p>IE is an indolent disease with a relevant thrombotic risk, lower than polycythaemia vera but higher than the general population [<span>8</span>]. In other forms of erythrocytosis, specific mutations have been shown to affect thrombotic risk assessment [<span>11</span>], but to date the role of mutational status in vascular complications of patients with IE has not yet been evaluated. The impact of <i>HFE</i> mutation on thrombotic risk has been evaluated in other cohorts. <i>HFE</i> mutations, in particular the C282Y mutation, do not seem to worsen both arterial and venous thrombotic risk in patients with hereditary hemochromatosis [<span>12, 13</span>]. Furthermore, it has been reported that the presence of <i>HFE</i> mutations does not impact thrombotic risk in patients with hereditary thrombophilia related to the presence of factor V Leiden [<span>14, 15</span>].</p><p>Although high haematocrit is certainly one of the promoters of increased thrombotic risk in patients with IE compared to the general population [<span>8, 10</span>], our study suggests that the presence of <i>HFE</i> mutations may be an additional risk factor for thrombotic complications, which in our series leads to a more than four-fold increase in thrombotic risk. The mechanism behind this finding is unclear. Considering that patients with hereditary hemochromatosis do not appear to have an increased thrombotic risk [<span>12-15</span>], the combined effect of dysregulation of iron metabolism due to <i>HFE</i> mutation and increased haematocrit may play a role. To date, it seems premature to recommend, in daily clinical practice, the inclusion of screening for <i>HFE</i> mutations in the diagnostic work-up of patients with IE. However, our observations, if confirmed on larger series, may improve risk stratification in order to achieve a more tailored diagnostic and therapeutic approach in patients with IE.</p><p>Irene Bertozzi and Andrea Benetti conceived the study, collected clinical data and wrote the paper. Elisabetta Cosi, Cecilia Fortino and Martina Zerbinati collected clinical data. Maria Luigia Randi and Paolo Simioni conceived the study, supervised research activities and wrote the paper.</p><p>The authors declare no conflicts of interest.</p><p>The authors received no specific funding for this work.</p><p>Informed consent has been obtained from patients included in the study.</p>\",\"PeriodicalId\":72883,\"journal\":{\"name\":\"EJHaem\",\"volume\":\"5 5\",\"pages\":\"1086-1088\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-10-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jha2.1019\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"EJHaem\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jha2.1019\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"EJHaem","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jha2.1019","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Impact of HFE mutations on thrombotic risk in patients with idiopathic erythrocytosis: A single-centre study
Idiopathic erythrocytosis (IE) is characterized by an increase in red blood cell mass without an identified cause. Diagnosis of IE is based on exclusion of all the known forms of primary and secondary acquired erythrocytosis and various congenital primary and secondary polycythaemias [1-3]. Recent studies have demonstrated a genetic complexity of IE, detecting the presence of several genetic variants in genes involved or suspected of being involved in erythrocytosis [4, 5]. In particular, HFE mutations are frequently observed in patients with IE, postulating that iron metabolism impairment is a possible underlying cause for erythrocytosis [6, 7].
IE shows a peculiar clinical phenotype (male, young, isolated erythrocytosis), a trend for a stable disease with no tendency to spontaneous progression to myelofibrosis or acute leukaemia, but a relevant risk of thrombosis, especially arterial events, also in young patients [5, 8, 9]. To date, no clear factors related to the increased thrombotic risk in IE have been established, therefore current therapeutic indications are aimed only at the management of cardiovascular risk factors. It has been shown that high haematocrit independently promotes arterial thrombosis by increasing the rate of platelet deposition and thrombus growth in spite of the absence of a clonal disease [10], but the role of mutational status in thrombotic risk assessment has never been explored in patients with IE.
We studied 100 patients referred to our department, with a diagnosis of IE and an available complete medical history, including common cardiovascular risk factors (hypertension, diabetes, dyslipidaemia and active smoking). None of them carried JAK2 V617F or exon 12 mutations [1]. Congenital primary and secondary polycythaemias were excluded in the absence of a familial pattern (i.e., at least one relative with erythrocytosis) and known mutations in EPO-R or Oxygen Sensing Pathway genes [2, 3]. A targeted next-generation sequencing (NGS) panel for patients with unexplained erythrocytosis was set up, including genes involved or suspected to be involved in erythrocytosis (Supporting information). Clinical and laboratory data of the patients are shown in Table 1.
All patients gave written informed consent. The protocol was approved by the local Institutional Ethical Committee (Azienda Ospedaliera di Padova, ref: 3922/AO/16). The study was conducted in compliance with the principles of the Declaration of Helsinki. The statistical tests adopted were logistic regression model for univariate and bivariate analysis and Cox regression model for survival analysis. Survival curve has been prepared with Kaplan–Meier method and compared with log rank test.
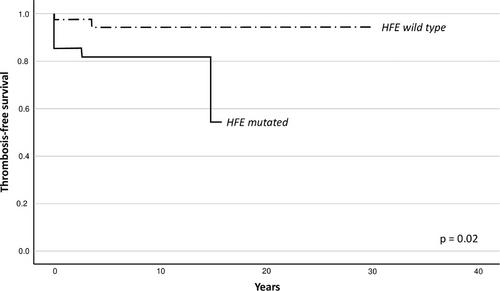
Sixty-seven (67%) patients carry at least one gene variant detected by the NGS study (Table S1). Forty-seven (47%) patients carry at least a mutation of HFE (30 heterozygous and 3 homozygous for H63D variant, 7 heterozygous for C282Y mutation, 2 heterozygous for C65S mutation, 3 compound heterozygous C282Y/H63D, 1 H63D/C65S and 1 C282Y/C65S). No differences in clinical and laboratory parameters have been found comparing patients with or without at least one gene variant detectable in the NGS study. Furthermore, among mutated patients no difference has been observed comparing clinical and laboratory parameters of the different detected variants. We observed 15 vascular events, 11 arterial (6 acute MI, 3 TIA, and 2 ischemic stroke) and 4 venous (2 DVT and 2 PE) thromboses in 13 patients (13%). In nine patients thrombotic event occurs at diagnosis or first evidence of erythrocytosis, in the other four during follow-up after a median time of 4.84 years. Total thrombosis rate in our cohort was 2.51 events*100 pats/years. In univariate analysis, patients with thrombotic complications were older (median age 64 vs. 56 y; p = 0.007, OR 1.071, CI 95% 1.02–1.13), had higher haematocrit (53 vs. 51%; p = 0.027, OR 1.31, CI 95% 1.03–1.66) and higher prevalence of HFE mutations [10 (77%) vs. 34 (39%), p = 0.028, OR 4.61, CI95% 1.18–18.02], while no difference has been reported in gender, haemoglobin and ferritin levels and prevalence of cardiovascular risk factors (Table S2). In bivariate analysis, both the presence of at least one HFE mutation (p = 0.043, OR 4.31, CI 95% 1.05–17.77) and older age at diagnosis (p = 0.011, OR 1.066, CI 95% 1.02–1.12) have been confirmed as risk factors for thrombosis. No difference was demonstrated in the occurrence of thrombotic complications comparing mutated and unmutated patients considering all detected variants in NGS, but stratifying patients on the basis of HFE mutational status (47 patients with at least one HFE mutation vs. 53 HFE wild-type patients) we found a significantly higher frequency of thrombotic complications in patients with at least one HFE mutation (n = 10, 21.3%) compared to all HFE wild-type patients (n = 3, 5.7%; p 0.03). Patients with and without HFE mutation had similar parameters at diagnosis and similar prevalence of cardiovascular risk factors. HFE mutated and wild-type patients showed an incidence of thrombosis of 4.4 and 1.28 events*100, respectively, with a relative risk of 3.44. Patients with HFE mutations showed a worse thrombosis-free survival compared to the wild-type patients (p 0.02; Figure 1), this observation was confirmed also in Cox regression analysis (p = 0.041, HR 5.04, CI 95% 1.07–23.80).
IE is an indolent disease with a relevant thrombotic risk, lower than polycythaemia vera but higher than the general population [8]. In other forms of erythrocytosis, specific mutations have been shown to affect thrombotic risk assessment [11], but to date the role of mutational status in vascular complications of patients with IE has not yet been evaluated. The impact of HFE mutation on thrombotic risk has been evaluated in other cohorts. HFE mutations, in particular the C282Y mutation, do not seem to worsen both arterial and venous thrombotic risk in patients with hereditary hemochromatosis [12, 13]. Furthermore, it has been reported that the presence of HFE mutations does not impact thrombotic risk in patients with hereditary thrombophilia related to the presence of factor V Leiden [14, 15].
Although high haematocrit is certainly one of the promoters of increased thrombotic risk in patients with IE compared to the general population [8, 10], our study suggests that the presence of HFE mutations may be an additional risk factor for thrombotic complications, which in our series leads to a more than four-fold increase in thrombotic risk. The mechanism behind this finding is unclear. Considering that patients with hereditary hemochromatosis do not appear to have an increased thrombotic risk [12-15], the combined effect of dysregulation of iron metabolism due to HFE mutation and increased haematocrit may play a role. To date, it seems premature to recommend, in daily clinical practice, the inclusion of screening for HFE mutations in the diagnostic work-up of patients with IE. However, our observations, if confirmed on larger series, may improve risk stratification in order to achieve a more tailored diagnostic and therapeutic approach in patients with IE.
Irene Bertozzi and Andrea Benetti conceived the study, collected clinical data and wrote the paper. Elisabetta Cosi, Cecilia Fortino and Martina Zerbinati collected clinical data. Maria Luigia Randi and Paolo Simioni conceived the study, supervised research activities and wrote the paper.
The authors declare no conflicts of interest.
The authors received no specific funding for this work.
Informed consent has been obtained from patients included in the study.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


