Ivo N. SahBandar, Gustavo H. B. Maegawa, Danielle Brandman, Jacob H. Rand, Hana I. Lim, Julia T. Geyer
{"title":"骨髓活检显示泡沫状组织细胞增生,诊断为非神经病性晚期酸性鞘磷脂酶缺乏症(尼曼-皮克病 A/B)。","authors":"Ivo N. SahBandar, Gustavo H. B. Maegawa, Danielle Brandman, Jacob H. Rand, Hana I. Lim, Julia T. Geyer","doi":"10.1002/jha2.1003","DOIUrl":null,"url":null,"abstract":"<p>The patient was a 44-year-old East-Asian descent male presenting with longstanding splenomegaly, thrombocytopenia, easy bruising since childhood, and cryptogenic cirrhosis with a history of recurrent variceal bleeding, currently being evaluated for a liver transplant. Pertinent laboratory findings include low serum albumin (3.6, <i>N</i> = 3.9–5.2 g/dL), increased total (4.8, <i>N</i> = 0.3–1.2 mg/dL), direct (1.3, <i>N</i> = ≤ 0.3 mg/dL), and indirect bilirubin (3.5, <i>N</i> = 0.1–0.8 mg/dL), mild AST elevation (53, <i>N</i> = ≤ 34 U/L), mild normocytic anemia (Hgb 119, <i>N</i> = 126–170 g/L, MCV 85.9, <i>N</i> = 78.6–94.2 fL), and thrombocytopenia (22 × 10e9, <i>N</i> = 156–325 × 10e9/L).</p><p>The bone marrow biopsy and clot section showed erythroid hyperplasia, decreased granulopoiesis with complete maturation for both lineages, and reduced megakaryocytes (Figure 1, panel A, H&E, 40X original magnification). A significant number of foamy histiocytes were identified, some of which contained cytoplasmic red blood cells (erythrophagocytosis, panel A). While most macrophages were negative for Periodic acid–Schiff (PAS) special stain, rare PAS-positive macrophages were seen (black arrow; Figure 1, panel B, PAS special stain, 40X original magnification). The described findings ruled out the presence of glycogen storage or Whipple disease. Representative macrophages (Figure 1, panels C–E, Wright-Giemsa, 100X original magnification) show typical features of acid sphingomyelinase deficiency (ASMD, aka Niemann-Pick disease types A/B) with low nuclear to cytoplasmic ratio and ample uniformly finely vacuolated cytoplasm, some contained red blood cells and debris (panel D), and sea blue histiocytes with deeply basophilic cytoplasm were noted (panel E). No organisms were identified by Grocott-Gomori methenamine silver (GMS) staining, and immunohistochemistry studies showed foamy histocytes non-reactivity to S100, langerin, and BRAF (figures not shown).</p><p>The clinical history and bone marrow biopsy findings were suspicious for lysosomal storage disease, and subsequent sphingomyelinase enzymatic activity (dried-blot spot, DBS) showed decreased residual activity (0.4 nmol/L, N ≥ 2.5 nmol/L) consistent with the late-onset non-neuronopathic form of ASMD, which is characterized by the development of hepatosplenomegaly and associated thrombocytopenia. In addition, the oxysterol, cholestane-3beta,5alpha- 6beta-triol (1.0 nmol/mL, <i>N</i> ≤ 0.8), and lyso-sphingomyelin (0.582 nmol/mL <i>N</i> ≤ 0.100) were elevated in DBS. Interestingly, chitotriosidase (492 nmols/h/mL, <i>N</i> 4–120) and angiotensin-converting enzyme (114 IU/L, <i>N</i> 16–85) were also elevated, reflecting the expanded reticulum endothelial system. Other lysosomal enzymes were at normal levels, as well as other sphingolipids, including lyso-glucosylphingosine. The case illustrates the importance of identifying bone marrow lipid-laden foam cells, triggering investigations for ASMD, to which Food and Drug Administration-approved disease-modifying therapy olipudase alpha (Xenpozyme), which can significantly improve the patient's splenomegaly, is currently available.</p><p>Ivo N. SahBandar and Julia T. Geyer designed the study, Ivo N. SahBandar, Julia T. Geyer, Gustavo H. B. Maegawa, and Jacob H. Rand conceived and analyzed the clinical and histologic data, Danielle Brandman and Hana I. Lim provided clinical data, Ivo N. SahBandar and Julia T. Geyer prepared the manuscript, Ivo N. SahBandar, Julia T. Geyer, Gustavo H. B. Maegawa, Hana I. Lim, Danielle Brandman and Jacob H. Rand edited the manuscript.</p><p>The authors declare no conflict of interest.</p><p>The authors received no specific funding for this work.</p><p>This study protocol was reviewed and approved by the Weill Cornell Medicine Institutional Review Board (WCM-IRB) at Weill Cornell Medical College of Cornell University, approval number 0107004999.</p><p>Written informed consent was obtained from the patient for publication of the details of their medical case and any accompanying images. Only deidentified data were used in this manuscript, and no information revealing the patient's identity was included. The New York Presbyterian Hospital/Weill Cornell Medicine is in compliance with the CARE guidelines for case reports.</p><p>The authors have confirmed clinical trial registration is not needed for this submission.</p>","PeriodicalId":72883,"journal":{"name":"EJHaem","volume":"5 5","pages":"1078-1079"},"PeriodicalIF":0.0000,"publicationDate":"2024-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jha2.1003","citationCount":"0","resultStr":"{\"title\":\"A diagnosis of non-neuronopathic and late-onset acid sphingomyelinase deficiency (Niemann-Pick disease A/B) following bone marrow biopsy showing foamy histiocytosis\",\"authors\":\"Ivo N. SahBandar, Gustavo H. B. Maegawa, Danielle Brandman, Jacob H. Rand, Hana I. Lim, Julia T. Geyer\",\"doi\":\"10.1002/jha2.1003\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The patient was a 44-year-old East-Asian descent male presenting with longstanding splenomegaly, thrombocytopenia, easy bruising since childhood, and cryptogenic cirrhosis with a history of recurrent variceal bleeding, currently being evaluated for a liver transplant. Pertinent laboratory findings include low serum albumin (3.6, <i>N</i> = 3.9–5.2 g/dL), increased total (4.8, <i>N</i> = 0.3–1.2 mg/dL), direct (1.3, <i>N</i> = ≤ 0.3 mg/dL), and indirect bilirubin (3.5, <i>N</i> = 0.1–0.8 mg/dL), mild AST elevation (53, <i>N</i> = ≤ 34 U/L), mild normocytic anemia (Hgb 119, <i>N</i> = 126–170 g/L, MCV 85.9, <i>N</i> = 78.6–94.2 fL), and thrombocytopenia (22 × 10e9, <i>N</i> = 156–325 × 10e9/L).</p><p>The bone marrow biopsy and clot section showed erythroid hyperplasia, decreased granulopoiesis with complete maturation for both lineages, and reduced megakaryocytes (Figure 1, panel A, H&E, 40X original magnification). A significant number of foamy histiocytes were identified, some of which contained cytoplasmic red blood cells (erythrophagocytosis, panel A). While most macrophages were negative for Periodic acid–Schiff (PAS) special stain, rare PAS-positive macrophages were seen (black arrow; Figure 1, panel B, PAS special stain, 40X original magnification). The described findings ruled out the presence of glycogen storage or Whipple disease. Representative macrophages (Figure 1, panels C–E, Wright-Giemsa, 100X original magnification) show typical features of acid sphingomyelinase deficiency (ASMD, aka Niemann-Pick disease types A/B) with low nuclear to cytoplasmic ratio and ample uniformly finely vacuolated cytoplasm, some contained red blood cells and debris (panel D), and sea blue histiocytes with deeply basophilic cytoplasm were noted (panel E). No organisms were identified by Grocott-Gomori methenamine silver (GMS) staining, and immunohistochemistry studies showed foamy histocytes non-reactivity to S100, langerin, and BRAF (figures not shown).</p><p>The clinical history and bone marrow biopsy findings were suspicious for lysosomal storage disease, and subsequent sphingomyelinase enzymatic activity (dried-blot spot, DBS) showed decreased residual activity (0.4 nmol/L, N ≥ 2.5 nmol/L) consistent with the late-onset non-neuronopathic form of ASMD, which is characterized by the development of hepatosplenomegaly and associated thrombocytopenia. In addition, the oxysterol, cholestane-3beta,5alpha- 6beta-triol (1.0 nmol/mL, <i>N</i> ≤ 0.8), and lyso-sphingomyelin (0.582 nmol/mL <i>N</i> ≤ 0.100) were elevated in DBS. Interestingly, chitotriosidase (492 nmols/h/mL, <i>N</i> 4–120) and angiotensin-converting enzyme (114 IU/L, <i>N</i> 16–85) were also elevated, reflecting the expanded reticulum endothelial system. Other lysosomal enzymes were at normal levels, as well as other sphingolipids, including lyso-glucosylphingosine. The case illustrates the importance of identifying bone marrow lipid-laden foam cells, triggering investigations for ASMD, to which Food and Drug Administration-approved disease-modifying therapy olipudase alpha (Xenpozyme), which can significantly improve the patient's splenomegaly, is currently available.</p><p>Ivo N. SahBandar and Julia T. Geyer designed the study, Ivo N. SahBandar, Julia T. Geyer, Gustavo H. B. Maegawa, and Jacob H. Rand conceived and analyzed the clinical and histologic data, Danielle Brandman and Hana I. Lim provided clinical data, Ivo N. SahBandar and Julia T. Geyer prepared the manuscript, Ivo N. SahBandar, Julia T. Geyer, Gustavo H. B. Maegawa, Hana I. Lim, Danielle Brandman and Jacob H. Rand edited the manuscript.</p><p>The authors declare no conflict of interest.</p><p>The authors received no specific funding for this work.</p><p>This study protocol was reviewed and approved by the Weill Cornell Medicine Institutional Review Board (WCM-IRB) at Weill Cornell Medical College of Cornell University, approval number 0107004999.</p><p>Written informed consent was obtained from the patient for publication of the details of their medical case and any accompanying images. Only deidentified data were used in this manuscript, and no information revealing the patient's identity was included. The New York Presbyterian Hospital/Weill Cornell Medicine is in compliance with the CARE guidelines for case reports.</p><p>The authors have confirmed clinical trial registration is not needed for this submission.</p>\",\"PeriodicalId\":72883,\"journal\":{\"name\":\"EJHaem\",\"volume\":\"5 5\",\"pages\":\"1078-1079\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-09-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jha2.1003\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"EJHaem\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jha2.1003\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"EJHaem","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jha2.1003","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
患者是一名 44 岁的东亚裔男性,长期脾肿大、血小板减少、自幼易淤血、隐源性肝硬化并有反复静脉曲张出血史,目前正在接受肝移植评估。相关实验室检查结果包括血清白蛋白偏低(3.6,N = 3.9-5.2 g/dL),总血清白蛋白(4.8,N = 0.3-1.2 mg/dL)、直接血清白蛋白(1.3,N = ≤ 0.3毫克/分升)、间接胆红素(3.5,N = 0.1-0.8 毫克/分升)、轻度谷草转氨酶升高(53,N = ≤ 34 U/L)、轻度正常红细胞性贫血(Hgb 119,N = 126-170 g/L,MCV 85.骨髓活检和血块切片显示红细胞增生,粒细胞生成减少,两系完全成熟,巨核细胞减少(图 1,A 面板,H&E,原始放大 40 倍)。发现大量泡沫组织细胞,其中一些含有细胞质红细胞(红细胞吞噬,A 组)。虽然大多数巨噬细胞的PAS(Periodic acid-Schiff)特异染色呈阴性,但也能看到极少数PAS阳性的巨噬细胞(黑色箭头;图1,B区,PAS特异染色,原始放大倍数40倍)。上述结果排除了糖原贮积症或 Whipple 病的可能。具有代表性的巨噬细胞(图1,C-E板,Wright-Giemsa,原始放大倍数100倍)显示出酸性鞘磷脂酶缺乏症(ASMD,又称尼曼-皮克病A/B型)的典型特征,细胞核与细胞质的比例较低,细胞质呈均匀细小的空泡状,其中一些含有红细胞和碎屑(D板),还发现了深嗜碱性细胞质的海蓝色组织细胞(E板)。Grocott-Gomori甲氰胺银(GMS)染色未发现生物体,免疫组化研究显示泡沫组织细胞对S100、langerin和BRAF无反应(图中未显示)。临床病史和骨髓活检结果均怀疑为溶酶体贮积病,随后的鞘磷脂酶活性(干印迹点,DBS)显示残留活性降低(0.4 nmol/L,N ≥ 2.5 nmol/L),这与晚发性非神经病变型 ASMD 一致,后者的特点是肝脾肿大和相关血小板减少。此外,氧甾醇、胆甾烷-3beta,5alpha- 6beta-三醇(1.0 nmol/mL,N ≤ 0.8)和溶血磷脂(0.582 nmol/mL,N ≤ 0.100)在 DBS 中升高。有趣的是,壳三糖苷酶(492 nmols/h/mL,N 4-120)和血管紧张素转换酶(114 IU/L,N 16-85)也升高了,这反映了网状内皮系统的扩大。其他溶酶体酶以及其他鞘磷脂(包括溶血葡萄糖鞘磷脂)均处于正常水平。该病例说明了识别骨髓脂质泡沫细胞的重要性,从而引发了对ASMD的检查,目前该病例已获得美国食品和药物管理局批准的疾病改变疗法olipudase alpha(Xenpozyme),该疗法可显著改善患者的脾肿大。Ivo N. SahBandar、Julia T. Geyer、Gustavo H. B. Maegawa和Jacob H. Rand构思并分析了临床和组织学数据,Danielle Brandman和Hana I. Lim提供了临床数据,Ivo N. SahBandar和Julia T. Geyer准备了手稿,Ivo N. SahBandar、Julia T. Geyer、Gustavo H. B. Maegawa、Hana I. Lim、Danielle Brandman和Jacob H. Rand编辑了手稿。本研究方案由康奈尔大学威尔康奈尔医学院的威尔康奈尔医学机构审查委员会(WCM-IRB)审查并批准,批准号为 0107004999。本研究已获得患者的书面知情同意,同意公布其医疗病例的细节和任何附带图片。本稿件仅使用了去身份化的数据,未包含任何暴露患者身份的信息。纽约长老会医院/威尔康奈尔医学院遵守CARE病例报告指南。作者已确认本稿件无需进行临床试验注册。
A diagnosis of non-neuronopathic and late-onset acid sphingomyelinase deficiency (Niemann-Pick disease A/B) following bone marrow biopsy showing foamy histiocytosis
The patient was a 44-year-old East-Asian descent male presenting with longstanding splenomegaly, thrombocytopenia, easy bruising since childhood, and cryptogenic cirrhosis with a history of recurrent variceal bleeding, currently being evaluated for a liver transplant. Pertinent laboratory findings include low serum albumin (3.6, N = 3.9–5.2 g/dL), increased total (4.8, N = 0.3–1.2 mg/dL), direct (1.3, N = ≤ 0.3 mg/dL), and indirect bilirubin (3.5, N = 0.1–0.8 mg/dL), mild AST elevation (53, N = ≤ 34 U/L), mild normocytic anemia (Hgb 119, N = 126–170 g/L, MCV 85.9, N = 78.6–94.2 fL), and thrombocytopenia (22 × 10e9, N = 156–325 × 10e9/L).
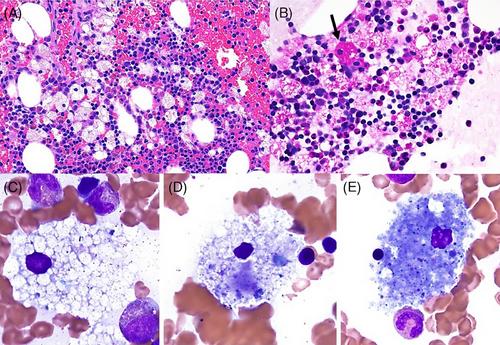
The bone marrow biopsy and clot section showed erythroid hyperplasia, decreased granulopoiesis with complete maturation for both lineages, and reduced megakaryocytes (Figure 1, panel A, H&E, 40X original magnification). A significant number of foamy histiocytes were identified, some of which contained cytoplasmic red blood cells (erythrophagocytosis, panel A). While most macrophages were negative for Periodic acid–Schiff (PAS) special stain, rare PAS-positive macrophages were seen (black arrow; Figure 1, panel B, PAS special stain, 40X original magnification). The described findings ruled out the presence of glycogen storage or Whipple disease. Representative macrophages (Figure 1, panels C–E, Wright-Giemsa, 100X original magnification) show typical features of acid sphingomyelinase deficiency (ASMD, aka Niemann-Pick disease types A/B) with low nuclear to cytoplasmic ratio and ample uniformly finely vacuolated cytoplasm, some contained red blood cells and debris (panel D), and sea blue histiocytes with deeply basophilic cytoplasm were noted (panel E). No organisms were identified by Grocott-Gomori methenamine silver (GMS) staining, and immunohistochemistry studies showed foamy histocytes non-reactivity to S100, langerin, and BRAF (figures not shown).
The clinical history and bone marrow biopsy findings were suspicious for lysosomal storage disease, and subsequent sphingomyelinase enzymatic activity (dried-blot spot, DBS) showed decreased residual activity (0.4 nmol/L, N ≥ 2.5 nmol/L) consistent with the late-onset non-neuronopathic form of ASMD, which is characterized by the development of hepatosplenomegaly and associated thrombocytopenia. In addition, the oxysterol, cholestane-3beta,5alpha- 6beta-triol (1.0 nmol/mL, N ≤ 0.8), and lyso-sphingomyelin (0.582 nmol/mL N ≤ 0.100) were elevated in DBS. Interestingly, chitotriosidase (492 nmols/h/mL, N 4–120) and angiotensin-converting enzyme (114 IU/L, N 16–85) were also elevated, reflecting the expanded reticulum endothelial system. Other lysosomal enzymes were at normal levels, as well as other sphingolipids, including lyso-glucosylphingosine. The case illustrates the importance of identifying bone marrow lipid-laden foam cells, triggering investigations for ASMD, to which Food and Drug Administration-approved disease-modifying therapy olipudase alpha (Xenpozyme), which can significantly improve the patient's splenomegaly, is currently available.
Ivo N. SahBandar and Julia T. Geyer designed the study, Ivo N. SahBandar, Julia T. Geyer, Gustavo H. B. Maegawa, and Jacob H. Rand conceived and analyzed the clinical and histologic data, Danielle Brandman and Hana I. Lim provided clinical data, Ivo N. SahBandar and Julia T. Geyer prepared the manuscript, Ivo N. SahBandar, Julia T. Geyer, Gustavo H. B. Maegawa, Hana I. Lim, Danielle Brandman and Jacob H. Rand edited the manuscript.
The authors declare no conflict of interest.
The authors received no specific funding for this work.
This study protocol was reviewed and approved by the Weill Cornell Medicine Institutional Review Board (WCM-IRB) at Weill Cornell Medical College of Cornell University, approval number 0107004999.
Written informed consent was obtained from the patient for publication of the details of their medical case and any accompanying images. Only deidentified data were used in this manuscript, and no information revealing the patient's identity was included. The New York Presbyterian Hospital/Weill Cornell Medicine is in compliance with the CARE guidelines for case reports.
The authors have confirmed clinical trial registration is not needed for this submission.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


