采用主动学习策略的铜锡机器学习原子间势研究
IF 3.1
3区 材料科学
Q2 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
摘要
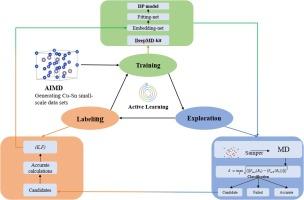
铜锡合金材料广泛应用于电子工业、航空航天和 3D 打印领域。在研究材料的结构和性能时,分子动力学(MD)会遇到算术和精度之间的矛盾。分子动力学是研究合金材料力学性能的一种通用理论计算方法。然而,由于传统原子间势的构建具有重复性和低效性,合金材料的分子动力学模拟仅限于简单体系。本研究利用深度神经网络模型构建了铜锡材料机器学习原子间势,并将第一性原理计算结果作为训练数据集,以确保量子力学原子间势的准确性。这一过程的特点是 "主动学习 "过程、数据生成和模型训练方法,人为干预极少。使用机器学习原子间势(MLIP)的分子动力学和第一原理计算显示出良好的一致性。结论是,MLIP 可以准确预测能量、力和机械性能。每个原子的能量和力的均方根误差(RMSE)约为 10 meV/原子和 100 meV/埃。它在能量-体积曲线、相变温度和弹性模量方面显示出良好的优势,为 MD 方法在铜锡合金材料设计和开发中的广泛应用奠定了基础。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Research on Cu-Sn machine learning interatomic potential with active learning strategy
Cu-Sn alloy materials are widely used in electronic industry, aerospace and 3D printing. When studying the structure and properties of materials, a contradiction between arithmetic and accuracy is encountered by Molecular dynamics (MD). Molecular dynamics is a general theoretical calculation method for studying the mechanical properties of alloy materials. However, molecular dynamics simulations of alloy materials are limited to simple systems because the construction of traditional interatomic potentials is replicative and inefficient. In this study, the Cu-Sn material machine learning interatomic potential was constructed by using a deep neural network model, and the first-principles calculation results were used as the training data set to ensure the accuracy of the quantum mechanical interatomic potential. This process features an “active learning” process, data generation and model training methods with minimal human intervention. Molecular dynamics using machine learning interatomic potentials (MLIP) and first-principles calculations show good consistency. It was concluded that MLIP can accurately predict energy, force and mechanical properties. The root mean square error (RMSEs) of the energy and force per atom is approximately 10 meV/atom and 100 meV/Å. It shows good advantages in energy-volume curve, phase transition temperature and elastic modulus, laying the foundation for the wide application of the MD method in the design and development of Cu-Sn alloy materials.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational Materials Science
工程技术-材料科学:综合
CiteScore
6.50
自引率
6.10%
发文量
665
审稿时长
26 days
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


