单壁碳纳米管解开 DNA 链:分子对接和 MD 模拟方法
IF 2.7
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
摘要
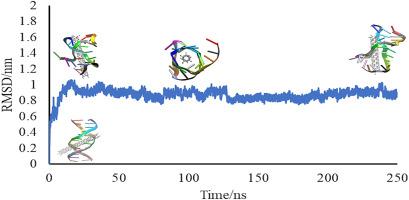
尽管有关碳纳米管(CNTs)在科学和医学领域应用的研究日益增多,但人们对其潜在毒性的担忧仍未得到充分研究。本研究利用分子对接计算结合分子动力学模拟来研究单壁碳纳米管(swCNTs)与双链 DNA(dsDNA)之间错综复杂的动态相互作用。通过研究 swCNT 特征(如长度、半径和手性)的影响,我们的研究结果揭示了形成 dsDNA-swCNT 复合物结合亲和力和稳定性的复杂相互作用。分子对接结果表明,半径为 0.16 Å、长度为 38 Å 的人字形 swCNT 与 dsDNA 的结合亲和力最高(-23.9 kcal/mol)。从对接结果、结合能、RMSD、回旋半径和平均力势(PMF)曲线等方面进行综合分析,可以详细了解变性动力学。PMF 曲线揭示了 DNA-CNT 相互作用的热力学可行性,勾勒出不同的能量景观和障碍:当选定的 swCNT 与 dsDNA 沟槽结合时,系统会被困在第一和第二个局部能量最小值处,分别为 1.48 nm 和 1.00 nm。分子内氢键计算结果表明,该效应显著降低,从而证实了 swCNT 对 DNA 的变性作用。此外,研究还发现,溴化乙锭(EB)与 swCNT 相互作用后,与 dsDNA 的结合亲和力显著降低,EB 与 dsDNA 的结合降低了约 13.2%。这项研究为了解 swCNT 对 dsDNA 的毒性作用提供了宝贵的见解,有助于合理解释 swCNT 的致癌潜力。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Unwinding DNA strands by single-walled carbon nanotubes: Molecular docking and MD simulation approach
Despite the growing research into the use of carbon nano-tubes (CNTs) in science and medicine, concerns about their potential toxicity remain insufficiently studied. This study utilizes molecular docking calculations combined by molecular dynamics simulations to investigate the dynamic intricacies of the interaction between single-walled carbon nanotubes (swCNTs) and double-stranded DNA (dsDNA). By examining the influence of swCNT characteristics such as length, radius, and chirality, our findings shed light on the complex interplay that shapes the binding affinity and stability of the dsDNA-swCNT complex. Molecular docking results identify a zigzag swCNT, with a radius of 0.16 Å and a length of 38 Å, as exhibiting the highest binding affinity with dsDNA (−23.9 kcal/mol). Comprehensive analyses, spanning docking results, binding energies, RMSD, radius of gyration, and potential of mean force (PMF) profiles, provide a detailed understanding of the denaturation dynamics. The PMF profiles reveal the thermodynamic feasibility of the DNA-CNT interaction, outlining distinct energy landscapes and barriers: when the selected swCNT binds within the dsDNA groove, the system becomes trapped at the first and second local energy minima, occurring at 1.48 nm and 1.00 nm, respectively. Intramolecular hydrogen bond calculations show a significant reduction, affirming the denaturing effect of swCNTs on DNA. Furthermore, the study reveals a significant reduction in the binding affinity of Ethidium Bromide (EB) to dsDNA following its interaction with swCNT, with a decrease in EB binding to dsDNA of approximately 13.2 %. This research offers valuable insights into the toxic effects of swCNTs on dsDNA, contributing to a rationalization of the cancerous potential of swCNTs.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


