铁-镍英达合金在高压下的氧化行为:ReaxFF 分子动力学研究
IF 3.1
3区 材料科学
Q2 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
摘要
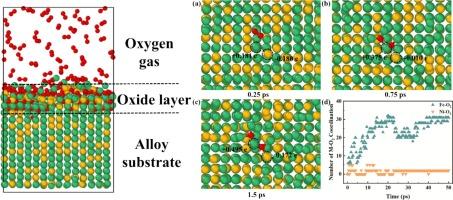
铁-镍英华尔合金经常在高温、高压和腐蚀性环境等极端条件下使用,因此很容易发生氧化,进而导致失效。因此,彻底了解其氧化行为至关重要。我们进行了基于 ReaxFF 的分子动力学(MD)模拟,以研究铁-镍因瓦合金在极端条件下的原子尺度氧化行为。氧化的初始阶段涉及 O2 的优先吸附和解离,表明了其表面-位点选择性。初始氧化动力学遵循对数规律,整个氧化过程导致氧化膜中的低配位簇构型占主导地位。与此同时,Fe 原子往往比 Ni 原子向 O 原子提供更多的电子。此外,还发现 O2 的消耗率随压力的增加而增加,并且在高压下更容易形成无定形氧化物。我们的研究结果表明,调节压力可增强铁-镍合金的抗氧化性,这对极端条件下合金的设计和应用具有重要意义。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Oxidation behavior of Fe-Ni Invar alloy under high pressure: A ReaxFF molecular dynamics study
The Fe-Ni Invar alloy is often employed under extreme conditions such as high temperatures, high pressures, and corrosive environments, making it susceptible to oxidation and subsequent failure. Hence, a thorough understanding of its oxidation behavior is crucial. We performed ReaxFF-based molecular dynamics (MD) simulations to study the oxidation behaviour of Fe-Ni Invar alloy at the atomic scale under extreme conditions. The initial stage of oxidation involves the preferential adsorption and dissociation of O2, demonstrating its surface-site selectivity. The initial oxidation kinetics follows a logarithmic law, and the whole oxidation process results in dominant low-coordinated clusters configurations in the oxide film. Simultaneously, Fe atoms tend to donate more electrons to O atoms than Ni atoms. Moreover, the O2 consumption rate were found to increase with pressure and amorphous oxides formed more readily under high pressure. Our results indicate that adjusting the pressure may enhance the oxidation resistance of the Fe-Ni alloy, which is significant for the design and application of alloys in extreme conditions.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational Materials Science
工程技术-材料科学:综合
CiteScore
6.50
自引率
6.10%
发文量
665
审稿时长
26 days
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


