Cs2(Ti, Zr, Hf)X6 双卤化物包晶带偏移的第一原理计算
IF 3.1
3区 材料科学
Q2 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
摘要
本研究探讨了 Cs2(Ti,Zr,Hf)X6 双卤化物包晶的能带偏移。研究采用密度泛函理论(DFT),使用 Perdew-Burke-Ernzerhof (PBE) 函数和混合 Heyd-Scuseria-Ernzerhof (HSE) 函数,利用超级电池技术计算了价带偏移(VBO)和导带偏移(CBO)。这种技术通过明确计算界面上的电位差,提供了更高的精度和可靠性。影响结果的一个关键因素是偶极电势(VD)的贡献,它导致 VBO 和 CBO 与电子亲和力规则预测值相比发生约 0.2 至 0.6 eV 的偏移。这种差异是由于加入了电荷再分布和极化等界面特异效应。此外,研究结果表明,这些化合物的能带排列在同一组卤化物中属于 I 型,由于离子半径相似,Zr- 和 Hf 基化合物的晶格常数几乎相同。这些结果为电子应用中异质结构的设计提供了宝贵的见解,并凸显了 Cs2(Ti、Zr、Hf)X6 化合物作为太阳能电池、发光二极管和光检测器的高效材料的潜力。本文章由计算机程序翻译,如有差异,请以英文原文为准。
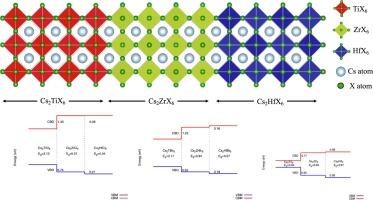
The first principle calculations of band offsets among Cs2(Ti, Zr, Hf)X6 double halide perovskites
This study investigates the band offsets among Cs2(Ti, Zr, Hf)X6 double halide perovskites. Valence band offsets (VBO) and conduction band offsets (CBO) were calculated using density functional theory (DFT) with the Perdew–Burke-Ernzerhof (PBE) functional and the hybrid Heyd–Scuseria–Ernzerhof (HSE) functional, employing the supercell technique. This technique offers greater accuracy and reliability by explicitly calculating the potential differences at the interface. A critical factor influencing the results is the contribution of the dipole potential (VD), which induces shifts in the VBO and CBO by approximately 0.2 to 0.6 eV relative to values predicted by the electron affinity rule. This discrepancy arises from the inclusion of interface-specific effects, such as charge redistribution and polarization. Additionally, the findings indicate that the energy band alignments among these compounds are type-I within the same group of halides, with nearly identical lattice constants for Zr- and Hf-based compounds due to their similar ionic radii. These results provide valuable insights for the design of heterostructures in electronic applications and highlight the potential of Cs2(Ti, Zr, Hf)X6 compounds as efficient materials for solar cells, light-emitting diodes, and photodetectors.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational Materials Science
工程技术-材料科学:综合
CiteScore
6.50
自引率
6.10%
发文量
665
审稿时长
26 days
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


