对苯并咪唑衍生物的结构、带隙能效应、电荷转移、反应性、热能和 NADPH 抑制活性的计算研究
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
这项工作包含对苯并咪唑衍生物的计算研究,包括苯并咪唑衍生物的密度泛函理论、电子结构和生物学评估。密度泛函理论评估从几何优化开始,然后是分子静电位、光谱分析、极化性研究以及通过频率计算进行的热力学分析。溶剂前沿分子轨道分析对分子性质的影响是通过 IEFPCM 溶解模型来模拟的。拓扑分析有助于确定分子的电子结构。生物学评估包括药代动力学特性评估和分子对接。药代动力学描述符是利用在线工具生成的,通过与药物相似性规则进行比较,并分析与分子的吸收、分布、代谢、排泄和毒性有关的描述符,评估了分子作为药物分子的功效。该分子与 7D3E 和 3A1F 这两个靶点的对接产生了良好的结合能,分别为 -7.39 和 -5.81 kcal/mol。本文章由计算机程序翻译,如有差异,请以英文原文为准。
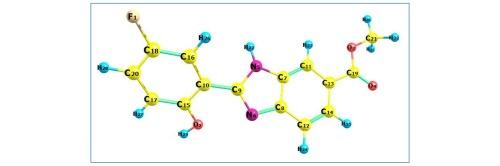
Computational investigation into the structure, effect of band gap energies, charge transfer, reactivity, thermal energies and NADPH inhibitory activity of a benzimidazole derivative
This work contains computational investigations of a benzimidazole derivative consisting of density functional theory, electronic structure and biological evaluation of a benzimidazole derivative. Density functional theory evaluation were conducted, starting from geometry optimisation, followed by the molecular electrostatic potential, spectral analyses, polarizability studies and thermodynamic analyses via the frequency calculations. Solvent frontier molecular orbital analyses, impact on the properties of the molecule were modelled with the IEFPCM model of solvation. Topological analyses helped to ascertain the molecule’s electronic structure. Biological assessment included pharmacokinetic property evaluation and molecular docking. Pharmacokinetic descriptors were generated using online tools and the molecule was assessed for its efficacy as a drug molecule by comparing with the rules concerning drug-likeness and analysing the descriptors relating to absorption, distribution, metabolism, excretion and toxicity of the molecule. Docking of the molecule with the two targets, 7D3E and 3A1F, yielded a good binding energy of −7.39 and −5.81 kcal/mol respectively.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


