对蛋白质二级结构独特特征的统计分析。
IF 2.6
4区 生物学
Q2 BIOLOGY
引用次数: 0
摘要
蛋白质折叠是一个受氨基酸主序列影响的复杂过程。早期的研究侧重于了解氨基酸的特异性或特性保持对于折叠成二级结构(如α螺旋、β片、转折和线圈)是否至关重要。然而,随着人工智能(AI)和机器学习(ML)的出现,重点已转向特定氨基酸的精确性质和出现。在我们的研究中,我们分析了来自不同生物体的大量蛋白质,以确定二级结构的独特特征,特别是极性、非极性和带电氨基酸残基的分布。我们发现,与其他二级结构相比,α-螺旋中带电和非极性基团的比例往往较高,螺旋中存在带相反电荷的氨基酸残基可使螺旋稳定,有利于形成较长的螺旋。这些特征与α螺旋截然不同。这项研究为蛋白质设计领域的研究人员提供了宝贵的见解,使他们能够重新创造短螺旋肽,用于一系列应用。我们还开发了一个网络服务器,用于广泛分析不同数据库中的蛋白质。网络服务器设在 https://proseqanalyser.iitgn.ac.in/。本文章由计算机程序翻译,如有差异,请以英文原文为准。
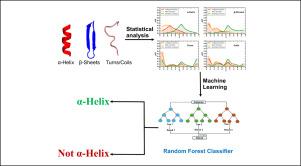
Statistical analysis of the unique characteristics of secondary structures in proteins
Protein folding is a complex process influenced by the primary sequence of amino acids. Early studies focused on understanding whether the specificity or the conservation of properties of amino acids was crucial for folding into secondary structures such as -helices, -sheets, turns, and coils. However, with the advent of artificial intelligence (AI) and machine learning (ML), the emphasis has shifted towards the precise nature and occurrence of specific amino acids. In our study, we analyzed a large set of proteins from diverse organisms to identify unique features of secondary structures, particularly in terms of the distribution of polar, non-polar, and charged amino acid residues. We found that -helices tend to have a higher proportion of charged and non-polar groups compared to other secondary structures and that the presence of oppositely charged amino acid residues in helices stabilizes them, facilitating the formation of longer helices. These characteristics are distinct to -helices. This study offers valuable insights for researchers in the field of protein design, enabling the de-novo creation of short helical peptides for a range of applications. We have also developed a web server for extensive analysis of proteins from different databases. The web server is housed at https://proseqanalyser.iitgn.ac.in/
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational Biology and Chemistry
生物-计算机:跨学科应用
CiteScore
6.10
自引率
3.20%
发文量
142
审稿时长
24 days
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


