威尔逊病中 ATP7B 剪接变体因与 COMMD1 的相互作用增强而导致功能障碍。
IF 7.1
1区 医学
Q1 GASTROENTEROLOGY & HEPATOLOGY
Cellular and Molecular Gastroenterology and Hepatology
Pub Date : 2024-10-09
DOI:10.1016/j.jcmgh.2024.101418
引用次数: 0
摘要
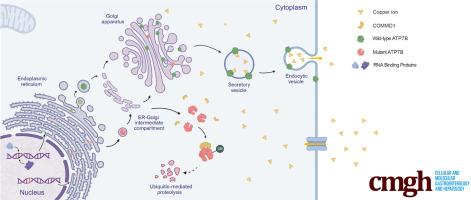
背景和目的:威尔森氏病与各种ATP7B突变之间的关联已被证实;然而,这些突变(尤其是剪接突变)的功能性后果的分子机制仍不清楚。本研究聚焦于ATP7B c.1543+1G>C变异,旨在揭示N端改变的ATP7B突变体的普遍致病机制:方法:通过剪接实验和RNA pull-down实验探讨剪接异常的机制。结果:c.1543+1G突变体对HuH-7细胞系和ATP7b-/-小鼠的功能影响进行了体外和体内评估:结果:c.1543+1G>C突变导致ATP7B第3外显子缺失,突变体ATP7B失去了跨高尔基网络(TGN)定位,并通过泛素-蛋白酶体途径降解,与COMMD1的相互作用增强。在表达突变型 ATP7B 的 HuH-7 细胞中观察到细胞间铜浓度升高和存活率降低。在Atp7b-/-小鼠体内恢复野生型ATP7B可显著改善表型,而用突变型ATP7B治疗的小鼠并没有表现出同等的益处:我们的研究调查了ATP7B c.1543+1G>C变体的致病性和机制,特别关注其与COMMD1的相互作用增强,这是导致各种ATP7B变体功能障碍的潜在普遍机制。这些发现为开发针对包括威尔逊病在内的一系列遗传性疾病中异常剪接事件的创新治疗策略奠定了基础。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Dysfunction of ATP7B Splicing Variant Caused by Enhanced Interaction With COMMD1 in Wilson Disease
Background & Aims
The association between Wilson disease and various ATP7B mutations is well-established; however, the molecular mechanism underlying the functional consequence of these mutations, particularly the splicing mutations, remains unclear. This study focused on the ATP7B c.1543+1G>C variant, to reveal a universal pathogenic mechanism of the ATP7B mutants with altered N-terminus.
Methods
The splicing assay and RNA pull-down were performed to explore the mechanism of the aberrant splicing. The ATP7B knockout HuH-7 cell line and Atp7b-/- mice were created, and the functional consequence of the mutant ATP7B were evaluated in vitro and in vivo.
Results
The c.1543+1G>C mutation resulted in the skipping of ATP7B exon 3, and the mutant ATP7B showed a loss of trans-Golgi network localization and was degraded via the ubiquitin-proteasome pathway, facilitated by enhanced interactions with COMMD1. Elevated intercellular copper concentration and reduced survival rate were observed in HuH-7 cells expressing mutant ATP7B. Restoration of wild-type ATP7B in Atp7b-/- mice resulted in a substantial improvement in phenotype, whereas mice treated with mutant ATP7B did not demonstrate equivalent benefits.
Conclusion
Our research investigated the pathogenicity and mechanism of ATP7B c.1543+1G>C variant, with particular focus on its enhanced interaction with COMMD1 as a potential universal mechanism contributing to the dysfunction of various ATP7B variants. These findings provide a foundation for the development of innovative therapeutic strategies that target abnormal splicing events in a range of hereditary diseases, including Wilson disease.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Cellular and Molecular Gastroenterology and Hepatology
Medicine-Gastroenterology
CiteScore
13.00
自引率
2.80%
发文量
246
审稿时长
42 days
期刊介绍:
"Cell and Molecular Gastroenterology and Hepatology (CMGH)" is a journal dedicated to advancing the understanding of digestive biology through impactful research that spans the spectrum of normal gastrointestinal, hepatic, and pancreatic functions, as well as their pathologies. The journal's mission is to publish high-quality, hypothesis-driven studies that offer mechanistic novelty and are methodologically robust, covering a wide range of themes in gastroenterology, hepatology, and pancreatology.
CMGH reports on the latest scientific advances in cell biology, immunology, physiology, microbiology, genetics, and neurobiology related to gastrointestinal, hepatobiliary, and pancreatic health and disease. The research published in CMGH is designed to address significant questions in the field, utilizing a variety of experimental approaches, including in vitro models, patient-derived tissues or cells, and animal models. This multifaceted approach enables the journal to contribute to both fundamental discoveries and their translation into clinical applications, ultimately aiming to improve patient care and treatment outcomes in digestive health.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


