{"title":"基于多叉 Logistic 回归模型的重叠组筛选过程的多类生存结果分类,并应用于 TCGA 转录组数据。","authors":"Jie-Huei Wang, Po-Lin Hou, Yi-Hau Chen","doi":"10.1177/11769351241286710","DOIUrl":null,"url":null,"abstract":"<p><strong>Objectives: </strong>Under the classification of multicategory survival outcomes of cancer patients, it is crucial to identify biomarkers that affect specific outcome categories. The classification of multicategory survival outcomes from transcriptomic data has been thoroughly investigated in computational biology. Nevertheless, several challenges must be addressed, including the ultra-high-dimensional feature space, feature contamination, and data imbalance, all of which contribute to the instability of the diagnostic model. Furthermore, although most methods achieve accurate predicted performance for binary classification with high-dimensional transcriptomic data, their extension to multi-class classification is not straightforward.</p><p><strong>Methods: </strong>We employ the One-versus-One strategy to transform multi-class classification into multiple binary classification, and utilize the overlapping group screening procedure with binary logistic regression to include pathway information for identifying important genes and gene-gene interactions for multicategory survival outcomes.</p><p><strong>Results: </strong>A series of simulation studies are conducted to compare the classification accuracy of our proposed approach with some existing machine learning methods. In practical data applications, we utilize the random oversampling procedure to tackle class imbalance issues. We then apply the proposed method to analyze transcriptomic data from various cancers in The Cancer Genome Atlas, such as kidney renal papillary cell carcinoma, lung adenocarcinoma, and head and neck squamous cell carcinoma. Our aim is to establish an accurate microarray-based multicategory cancer diagnosis model. The numerical results illustrate that the new proposal effectively enhances cancer diagnosis compared to approaches that neglect pathway information.</p><p><strong>Conclusions: </strong>We showcase the effectiveness of the proposed method in terms of class prediction accuracy through evaluations on simulated synthetic datasets as well as real dataset applications. We also identified the cancer-related gene-gene interaction biomarkers and reported the corresponding network structure. According to the identified major genes and gene-gene interactions, we can predict for each patient the probabilities that he/she belongs to each of the survival outcome classes.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":"23 ","pages":"11769351241286710"},"PeriodicalIF":2.5000,"publicationDate":"2024-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11462568/pdf/","citationCount":"0","resultStr":"{\"title\":\"Multicategory Survival Outcomes Classification via Overlapping Group Screening Process Based on Multinomial Logistic Regression Model With Application to TCGA Transcriptomic Data.\",\"authors\":\"Jie-Huei Wang, Po-Lin Hou, Yi-Hau Chen\",\"doi\":\"10.1177/11769351241286710\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objectives: </strong>Under the classification of multicategory survival outcomes of cancer patients, it is crucial to identify biomarkers that affect specific outcome categories. The classification of multicategory survival outcomes from transcriptomic data has been thoroughly investigated in computational biology. Nevertheless, several challenges must be addressed, including the ultra-high-dimensional feature space, feature contamination, and data imbalance, all of which contribute to the instability of the diagnostic model. Furthermore, although most methods achieve accurate predicted performance for binary classification with high-dimensional transcriptomic data, their extension to multi-class classification is not straightforward.</p><p><strong>Methods: </strong>We employ the One-versus-One strategy to transform multi-class classification into multiple binary classification, and utilize the overlapping group screening procedure with binary logistic regression to include pathway information for identifying important genes and gene-gene interactions for multicategory survival outcomes.</p><p><strong>Results: </strong>A series of simulation studies are conducted to compare the classification accuracy of our proposed approach with some existing machine learning methods. In practical data applications, we utilize the random oversampling procedure to tackle class imbalance issues. We then apply the proposed method to analyze transcriptomic data from various cancers in The Cancer Genome Atlas, such as kidney renal papillary cell carcinoma, lung adenocarcinoma, and head and neck squamous cell carcinoma. Our aim is to establish an accurate microarray-based multicategory cancer diagnosis model. The numerical results illustrate that the new proposal effectively enhances cancer diagnosis compared to approaches that neglect pathway information.</p><p><strong>Conclusions: </strong>We showcase the effectiveness of the proposed method in terms of class prediction accuracy through evaluations on simulated synthetic datasets as well as real dataset applications. We also identified the cancer-related gene-gene interaction biomarkers and reported the corresponding network structure. According to the identified major genes and gene-gene interactions, we can predict for each patient the probabilities that he/she belongs to each of the survival outcome classes.</p>\",\"PeriodicalId\":35418,\"journal\":{\"name\":\"Cancer Informatics\",\"volume\":\"23 \",\"pages\":\"11769351241286710\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-10-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11462568/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cancer Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11769351241286710\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351241286710","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Multicategory Survival Outcomes Classification via Overlapping Group Screening Process Based on Multinomial Logistic Regression Model With Application to TCGA Transcriptomic Data.
Objectives: Under the classification of multicategory survival outcomes of cancer patients, it is crucial to identify biomarkers that affect specific outcome categories. The classification of multicategory survival outcomes from transcriptomic data has been thoroughly investigated in computational biology. Nevertheless, several challenges must be addressed, including the ultra-high-dimensional feature space, feature contamination, and data imbalance, all of which contribute to the instability of the diagnostic model. Furthermore, although most methods achieve accurate predicted performance for binary classification with high-dimensional transcriptomic data, their extension to multi-class classification is not straightforward.
Methods: We employ the One-versus-One strategy to transform multi-class classification into multiple binary classification, and utilize the overlapping group screening procedure with binary logistic regression to include pathway information for identifying important genes and gene-gene interactions for multicategory survival outcomes.
Results: A series of simulation studies are conducted to compare the classification accuracy of our proposed approach with some existing machine learning methods. In practical data applications, we utilize the random oversampling procedure to tackle class imbalance issues. We then apply the proposed method to analyze transcriptomic data from various cancers in The Cancer Genome Atlas, such as kidney renal papillary cell carcinoma, lung adenocarcinoma, and head and neck squamous cell carcinoma. Our aim is to establish an accurate microarray-based multicategory cancer diagnosis model. The numerical results illustrate that the new proposal effectively enhances cancer diagnosis compared to approaches that neglect pathway information.
Conclusions: We showcase the effectiveness of the proposed method in terms of class prediction accuracy through evaluations on simulated synthetic datasets as well as real dataset applications. We also identified the cancer-related gene-gene interaction biomarkers and reported the corresponding network structure. According to the identified major genes and gene-gene interactions, we can predict for each patient the probabilities that he/she belongs to each of the survival outcome classes.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.
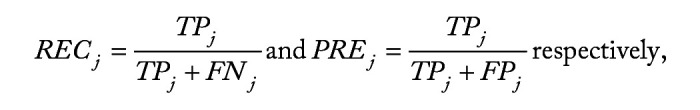
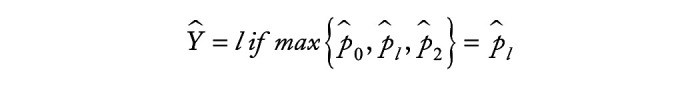
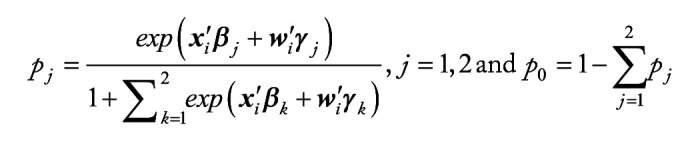

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


