Shuxian Xie, Chao Lv, Lichun Kong, Cui Li, Chang Wang, Xuyu Lv, Qianmin Wu, Jiuju Feng, Ai-Jun Wang, De-Li Chen and Fa Yang
{"title":"电位驱动原位形成富含 Se-空位的 CuS@Cu2Se 以引导从 HCOOH 到 C2H5OH 的 CO2 电还原路径","authors":"Shuxian Xie, Chao Lv, Lichun Kong, Cui Li, Chang Wang, Xuyu Lv, Qianmin Wu, Jiuju Feng, Ai-Jun Wang, De-Li Chen and Fa Yang","doi":"10.1039/D4QI02076F","DOIUrl":null,"url":null,"abstract":"<p >Copper chalcogenides are susceptible to electrochemical reconstruction, thus posing challenges in understanding the precise structure–function relationships during the CO<small><sub>2</sub></small> electroreduction reaction (CO<small><sub>2</sub></small>RR). Here, we synthesize a hierarchical core–shell CuS@CuSe catalyst, exhibiting a controllable selectivity from 67.5% for HCOOH at −0.5 V <em>vs.</em> RHE to 54.7% for C<small><sub>2</sub></small>H<small><sub>5</sub></small>OH at −0.9 V <em>vs.</em> RHE. Overlap-labeled transmission electron microscopy and <em>in situ</em> Raman spectroscopy dynamically monitor the potential-dependent structural evolution from the pristine CuS@CuSe to CuS@Cu<small><sub>2</sub></small>Se with Se vacancies (Cu<small><sub>2</sub></small>Se-<em>V</em><small><sub>Se</sub></small>). Density functional theory (DFT) calculations reveal that the generated Se-vacancies stabilize Cu<small><sup>+</sup></small> sites with shortened Cu–Cu spacing of 2.46 Å. This not only increases the affinities to the adsorbed *COOH and *CO species but also promotes the easier dimerization of *CO to form *OCCO (Δ<em>G</em> ∼ −0.50 eV) while suppressing its direct desorption to CO (Δ<em>G</em> ∼ +1.63 eV) or hydrogenation to *CHO (Δ<em>G</em> ∼ +0.74 eV) and *COH (Δ<em>G</em> ∼ +1.15 eV). This is believed to determine the remarkable ethanol selectivity. Furthermore, the rapid dissociation of water over the synergistic CuS sites kinetically accelerates the proton-coupling process. Such potential-dependent imperative intermediates associated with the bifurcated pathway are directly distinguished by isotope labelling <em>in situ</em> infrared spectroscopy. This work provides insights into designing an electrochemical reconstructed copper chalcogenide catalyst for tuning the C<small><sub>1</sub></small>/C<small><sub>2</sub></small> product selectivity in CO<small><sub>2</sub></small>RR technology.</p>","PeriodicalId":79,"journal":{"name":"Inorganic Chemistry Frontiers","volume":" 23","pages":" 8272-8284"},"PeriodicalIF":6.1000,"publicationDate":"2024-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Potential-driven in situ formation of Se-vacancy-rich CuS@Cu2Se to steer the CO2 electroreduction path from HCOOH to C2H5OH†\",\"authors\":\"Shuxian Xie, Chao Lv, Lichun Kong, Cui Li, Chang Wang, Xuyu Lv, Qianmin Wu, Jiuju Feng, Ai-Jun Wang, De-Li Chen and Fa Yang\",\"doi\":\"10.1039/D4QI02076F\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Copper chalcogenides are susceptible to electrochemical reconstruction, thus posing challenges in understanding the precise structure–function relationships during the CO<small><sub>2</sub></small> electroreduction reaction (CO<small><sub>2</sub></small>RR). Here, we synthesize a hierarchical core–shell CuS@CuSe catalyst, exhibiting a controllable selectivity from 67.5% for HCOOH at −0.5 V <em>vs.</em> RHE to 54.7% for C<small><sub>2</sub></small>H<small><sub>5</sub></small>OH at −0.9 V <em>vs.</em> RHE. Overlap-labeled transmission electron microscopy and <em>in situ</em> Raman spectroscopy dynamically monitor the potential-dependent structural evolution from the pristine CuS@CuSe to CuS@Cu<small><sub>2</sub></small>Se with Se vacancies (Cu<small><sub>2</sub></small>Se-<em>V</em><small><sub>Se</sub></small>). Density functional theory (DFT) calculations reveal that the generated Se-vacancies stabilize Cu<small><sup>+</sup></small> sites with shortened Cu–Cu spacing of 2.46 Å. This not only increases the affinities to the adsorbed *COOH and *CO species but also promotes the easier dimerization of *CO to form *OCCO (Δ<em>G</em> ∼ −0.50 eV) while suppressing its direct desorption to CO (Δ<em>G</em> ∼ +1.63 eV) or hydrogenation to *CHO (Δ<em>G</em> ∼ +0.74 eV) and *COH (Δ<em>G</em> ∼ +1.15 eV). This is believed to determine the remarkable ethanol selectivity. Furthermore, the rapid dissociation of water over the synergistic CuS sites kinetically accelerates the proton-coupling process. Such potential-dependent imperative intermediates associated with the bifurcated pathway are directly distinguished by isotope labelling <em>in situ</em> infrared spectroscopy. This work provides insights into designing an electrochemical reconstructed copper chalcogenide catalyst for tuning the C<small><sub>1</sub></small>/C<small><sub>2</sub></small> product selectivity in CO<small><sub>2</sub></small>RR technology.</p>\",\"PeriodicalId\":79,\"journal\":{\"name\":\"Inorganic Chemistry Frontiers\",\"volume\":\" 23\",\"pages\":\" 8272-8284\"},\"PeriodicalIF\":6.1000,\"publicationDate\":\"2024-10-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Inorganic Chemistry Frontiers\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/qi/d4qi02076f\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Inorganic Chemistry Frontiers","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/qi/d4qi02076f","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
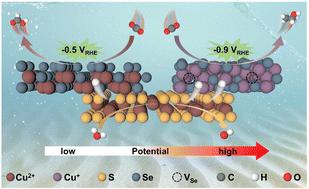
Potential-driven in situ formation of Se-vacancy-rich CuS@Cu2Se to steer the CO2 electroreduction path from HCOOH to C2H5OH†
Copper chalcogenides are susceptible to electrochemical reconstruction, thus posing challenges in understanding the precise structure–function relationships during the CO2 electroreduction reaction (CO2RR). Here, we synthesize a hierarchical core–shell CuS@CuSe catalyst, exhibiting a controllable selectivity from 67.5% for HCOOH at −0.5 V vs. RHE to 54.7% for C2H5OH at −0.9 V vs. RHE. Overlap-labeled transmission electron microscopy and in situ Raman spectroscopy dynamically monitor the potential-dependent structural evolution from the pristine CuS@CuSe to CuS@Cu2Se with Se vacancies (Cu2Se-VSe). Density functional theory (DFT) calculations reveal that the generated Se-vacancies stabilize Cu+ sites with shortened Cu–Cu spacing of 2.46 Å. This not only increases the affinities to the adsorbed *COOH and *CO species but also promotes the easier dimerization of *CO to form *OCCO (ΔG ∼ −0.50 eV) while suppressing its direct desorption to CO (ΔG ∼ +1.63 eV) or hydrogenation to *CHO (ΔG ∼ +0.74 eV) and *COH (ΔG ∼ +1.15 eV). This is believed to determine the remarkable ethanol selectivity. Furthermore, the rapid dissociation of water over the synergistic CuS sites kinetically accelerates the proton-coupling process. Such potential-dependent imperative intermediates associated with the bifurcated pathway are directly distinguished by isotope labelling in situ infrared spectroscopy. This work provides insights into designing an electrochemical reconstructed copper chalcogenide catalyst for tuning the C1/C2 product selectivity in CO2RR technology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


