Lisa Asta, Arianna Ricciardello, Francesca Cucinotta, Laura Turriziani, Maria Boncoddo, Fabiana Bellomo, Jessica Angelini, Martina Gnazzo, Giulia Scandolo, Giulia Pisanò, Francesco Pelagatti, Fethia Chehbani, Michela Camia, Antonio M Persico
{"title":"对 70 名意大利菲兰-麦克德米综合征患者进行临床、发育和血清素血症表型分析。","authors":"Lisa Asta, Arianna Ricciardello, Francesca Cucinotta, Laura Turriziani, Maria Boncoddo, Fabiana Bellomo, Jessica Angelini, Martina Gnazzo, Giulia Scandolo, Giulia Pisanò, Francesco Pelagatti, Fethia Chehbani, Michela Camia, Antonio M Persico","doi":"10.1186/s11689-024-09572-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Phelan-McDermid syndrome (PMS) is caused by monoallelic loss or inactivation at the SHANK3 gene, located in human chr 22q13.33, and is often associated with Autism Spectrum Disorder (ASD).</p><p><strong>Objectives: </strong>To assess the clinical and developmental phenotype in a novel sample of PMS patients, including for the first time auxometric trajectories and serotonin blood levels.</p><p><strong>Methods: </strong>70 Italian PMS patients were clinically characterized by parental report, direct medical observation, and a thorough medical and psychodiagnostic protocol. Serotonin levels were measured in platelet-rich plasma by HPLC.</p><p><strong>Results: </strong>Our sample includes 59 (84.3%) cases with chr. 22q13 terminal deletion, 5 (7.1%) disruptive SHANK3 mutations, and 6 (8.6%) ring chromosome 22. Intellectual disability was present in 69 (98.6%) cases, motor coordination disorder in 65 (92.9%), ASD in 20 (28.6%), and lifetime bipolar disorder in 12 (17.1%). Prenatal and postnatal complications were frequent (22.9%-48.6%). Expressive and receptive language were absent in 49 (70.0%) and 19 (27.1%) cases, respectively. Decreased pain sensitivity was reported in 56 (80.0%), hyperactivity in 49 (80.3%), abnormal sleep in 45 (64.3%), congenital dysmorphisms in 35 (58.3%), chronic stool abnormalities and especially constipation in 29 (41.4%). Parents reported noticing behavioral abnormalities during early childhood immediately after an infective episode in 34 (48.6%) patients. Brain MRI anomalies were observed in 53 (79.1%), EEG abnormalities in 16 (23.5%), kidney and upper urinary tract malformations in 18 (28.1%). Two novel phenotypes emerged: (a) a subgroup of 12/44 (27.3%) PMS patients displays smaller head size at enrollment (mean age 11.8 yrs) compared to their first year of neonatal life, documenting a deceleration of head growth (p < 0.001); (b) serotonin blood levels are significantly lower in 21 PMS patients compared to their 21 unaffected siblings (P < 0.05), and to 432 idiopathic ASD cases (p < 0.001).</p><p><strong>Conclusions: </strong>We replicate and extend the description of many phenotypic characteristics present in PMS, and report two novel features: (1) growth trajectories are variable and head growth appears to slow down during childhood in some PMS patients; (2) serotonin blood levels are decreased in PMS, and not increased as frequently occurs in ASD. Further investigations of these novel features are under way.</p>","PeriodicalId":16530,"journal":{"name":"Journal of Neurodevelopmental Disorders","volume":"16 1","pages":"57"},"PeriodicalIF":4.0000,"publicationDate":"2024-10-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11451156/pdf/","citationCount":"0","resultStr":"{\"title\":\"Clinical, developmental and serotonemia phenotyping of a sample of 70 Italian patients with Phelan-McDermid Syndrome.\",\"authors\":\"Lisa Asta, Arianna Ricciardello, Francesca Cucinotta, Laura Turriziani, Maria Boncoddo, Fabiana Bellomo, Jessica Angelini, Martina Gnazzo, Giulia Scandolo, Giulia Pisanò, Francesco Pelagatti, Fethia Chehbani, Michela Camia, Antonio M Persico\",\"doi\":\"10.1186/s11689-024-09572-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Phelan-McDermid syndrome (PMS) is caused by monoallelic loss or inactivation at the SHANK3 gene, located in human chr 22q13.33, and is often associated with Autism Spectrum Disorder (ASD).</p><p><strong>Objectives: </strong>To assess the clinical and developmental phenotype in a novel sample of PMS patients, including for the first time auxometric trajectories and serotonin blood levels.</p><p><strong>Methods: </strong>70 Italian PMS patients were clinically characterized by parental report, direct medical observation, and a thorough medical and psychodiagnostic protocol. Serotonin levels were measured in platelet-rich plasma by HPLC.</p><p><strong>Results: </strong>Our sample includes 59 (84.3%) cases with chr. 22q13 terminal deletion, 5 (7.1%) disruptive SHANK3 mutations, and 6 (8.6%) ring chromosome 22. Intellectual disability was present in 69 (98.6%) cases, motor coordination disorder in 65 (92.9%), ASD in 20 (28.6%), and lifetime bipolar disorder in 12 (17.1%). Prenatal and postnatal complications were frequent (22.9%-48.6%). Expressive and receptive language were absent in 49 (70.0%) and 19 (27.1%) cases, respectively. Decreased pain sensitivity was reported in 56 (80.0%), hyperactivity in 49 (80.3%), abnormal sleep in 45 (64.3%), congenital dysmorphisms in 35 (58.3%), chronic stool abnormalities and especially constipation in 29 (41.4%). Parents reported noticing behavioral abnormalities during early childhood immediately after an infective episode in 34 (48.6%) patients. Brain MRI anomalies were observed in 53 (79.1%), EEG abnormalities in 16 (23.5%), kidney and upper urinary tract malformations in 18 (28.1%). Two novel phenotypes emerged: (a) a subgroup of 12/44 (27.3%) PMS patients displays smaller head size at enrollment (mean age 11.8 yrs) compared to their first year of neonatal life, documenting a deceleration of head growth (p < 0.001); (b) serotonin blood levels are significantly lower in 21 PMS patients compared to their 21 unaffected siblings (P < 0.05), and to 432 idiopathic ASD cases (p < 0.001).</p><p><strong>Conclusions: </strong>We replicate and extend the description of many phenotypic characteristics present in PMS, and report two novel features: (1) growth trajectories are variable and head growth appears to slow down during childhood in some PMS patients; (2) serotonin blood levels are decreased in PMS, and not increased as frequently occurs in ASD. Further investigations of these novel features are under way.</p>\",\"PeriodicalId\":16530,\"journal\":{\"name\":\"Journal of Neurodevelopmental Disorders\",\"volume\":\"16 1\",\"pages\":\"57\"},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2024-10-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11451156/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Neurodevelopmental Disorders\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s11689-024-09572-7\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Neurodevelopmental Disorders","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s11689-024-09572-7","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Clinical, developmental and serotonemia phenotyping of a sample of 70 Italian patients with Phelan-McDermid Syndrome.
Background: Phelan-McDermid syndrome (PMS) is caused by monoallelic loss or inactivation at the SHANK3 gene, located in human chr 22q13.33, and is often associated with Autism Spectrum Disorder (ASD).
Objectives: To assess the clinical and developmental phenotype in a novel sample of PMS patients, including for the first time auxometric trajectories and serotonin blood levels.
Methods: 70 Italian PMS patients were clinically characterized by parental report, direct medical observation, and a thorough medical and psychodiagnostic protocol. Serotonin levels were measured in platelet-rich plasma by HPLC.
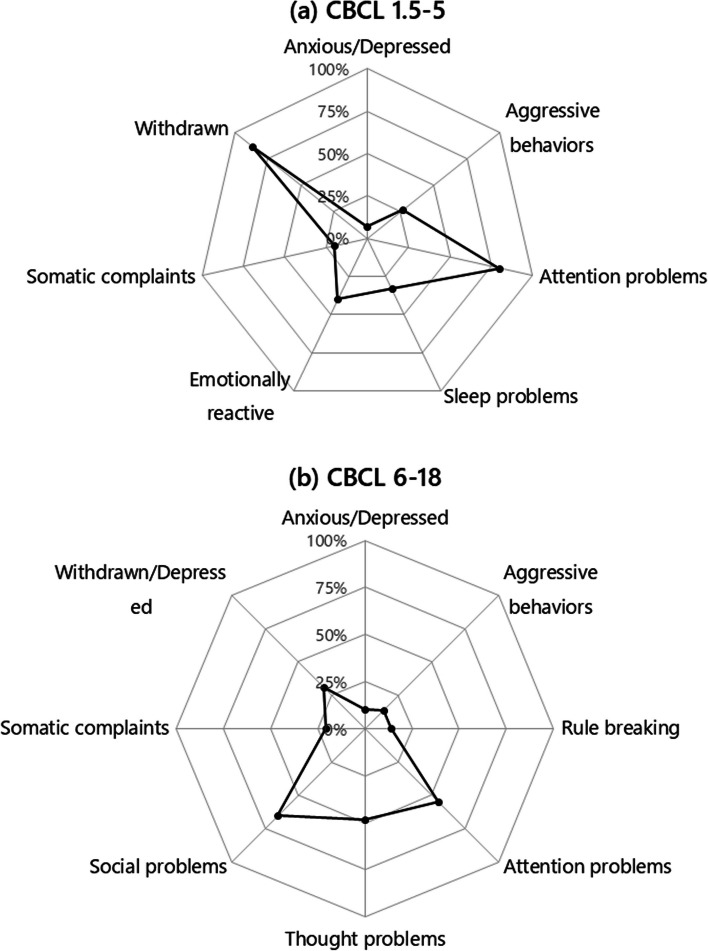
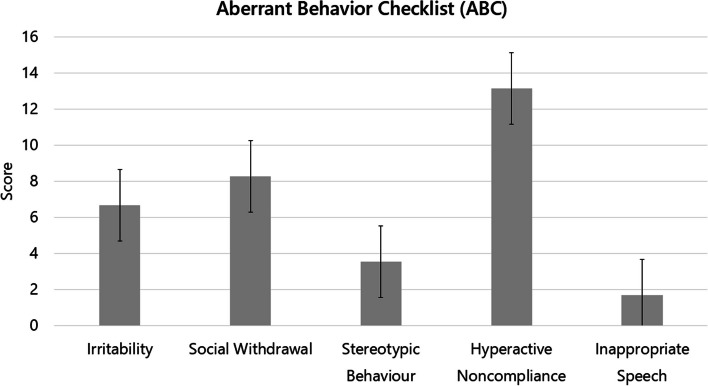
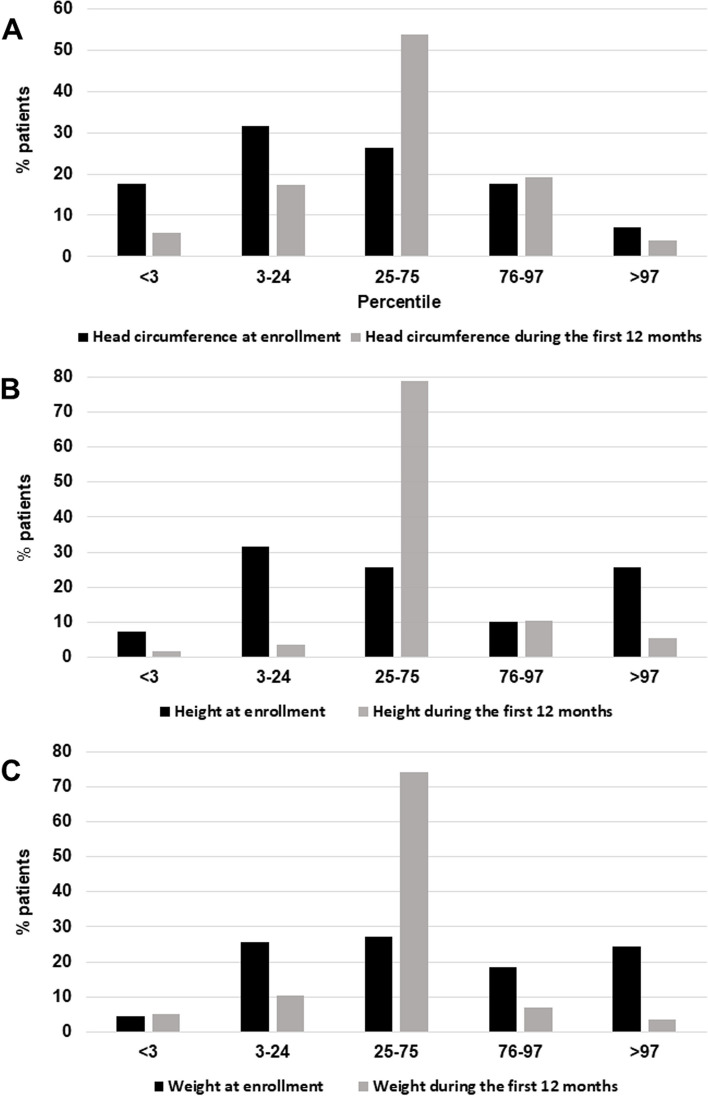
Results: Our sample includes 59 (84.3%) cases with chr. 22q13 terminal deletion, 5 (7.1%) disruptive SHANK3 mutations, and 6 (8.6%) ring chromosome 22. Intellectual disability was present in 69 (98.6%) cases, motor coordination disorder in 65 (92.9%), ASD in 20 (28.6%), and lifetime bipolar disorder in 12 (17.1%). Prenatal and postnatal complications were frequent (22.9%-48.6%). Expressive and receptive language were absent in 49 (70.0%) and 19 (27.1%) cases, respectively. Decreased pain sensitivity was reported in 56 (80.0%), hyperactivity in 49 (80.3%), abnormal sleep in 45 (64.3%), congenital dysmorphisms in 35 (58.3%), chronic stool abnormalities and especially constipation in 29 (41.4%). Parents reported noticing behavioral abnormalities during early childhood immediately after an infective episode in 34 (48.6%) patients. Brain MRI anomalies were observed in 53 (79.1%), EEG abnormalities in 16 (23.5%), kidney and upper urinary tract malformations in 18 (28.1%). Two novel phenotypes emerged: (a) a subgroup of 12/44 (27.3%) PMS patients displays smaller head size at enrollment (mean age 11.8 yrs) compared to their first year of neonatal life, documenting a deceleration of head growth (p < 0.001); (b) serotonin blood levels are significantly lower in 21 PMS patients compared to their 21 unaffected siblings (P < 0.05), and to 432 idiopathic ASD cases (p < 0.001).
Conclusions: We replicate and extend the description of many phenotypic characteristics present in PMS, and report two novel features: (1) growth trajectories are variable and head growth appears to slow down during childhood in some PMS patients; (2) serotonin blood levels are decreased in PMS, and not increased as frequently occurs in ASD. Further investigations of these novel features are under way.
期刊介绍:
Journal of Neurodevelopmental Disorders is an open access journal that integrates current, cutting-edge research across a number of disciplines, including neurobiology, genetics, cognitive neuroscience, psychiatry and psychology. The journal’s primary focus is on the pathogenesis of neurodevelopmental disorders including autism, fragile X syndrome, tuberous sclerosis, Turner Syndrome, 22q Deletion Syndrome, Prader-Willi and Angelman Syndrome, Williams syndrome, lysosomal storage diseases, dyslexia, specific language impairment and fetal alcohol syndrome. With the discovery of specific genes underlying neurodevelopmental syndromes, the emergence of powerful tools for studying neural circuitry, and the development of new approaches for exploring molecular mechanisms, interdisciplinary research on the pathogenesis of neurodevelopmental disorders is now increasingly common. Journal of Neurodevelopmental Disorders provides a unique venue for researchers interested in comparing and contrasting mechanisms and characteristics related to the pathogenesis of the full range of neurodevelopmental disorders, sharpening our understanding of the etiology and relevant phenotypes of each condition.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


