III-V 族半导体的电子和光学特性:砷化物和锑化物
IF 3.1
3区 材料科学
Q2 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
摘要
研究锌蓝III-V族半导体(尤其是砷化物和锑化物)的结构、电子和光学特性,由于它们具有广泛的直接带隙,因此对晶体管、红外探测器和量子技术等光电设备至关重要。在这项研究中,我们采用了第一原理方法,将 G0W0 与 HSE06 混合函数和自旋轨道耦合 (SOC) 结合起来,研究它们的基本特性。传统的密度泛函理论(DFT)方法,尤其是使用广义梯度逼近(GGA)PBE 函数的方法,往往会低估带隙,从而导致与实验结果的差异。为了解决这个问题,我们的研究纠正了带隙低估,并完善了光学常数的计算,包括介电函数、折射率、消光系数和吸收系数。此外,我们的计算模型得出的优化晶格常数和电子特性与实验数据密切相关,证明了模型在预测材料特性方面的可靠性。研究结果表明,我们的方法可应用于砷化物和锑化物,为设计具有光电特性的材料提供了一条途径,这些材料涉及 III-V 化合物及其复杂的异质结构,可用于先进的设备应用。本文章由计算机程序翻译,如有差异,请以英文原文为准。
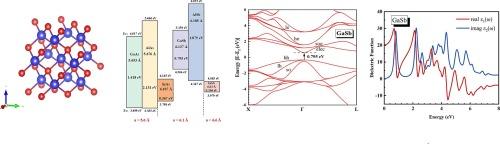
The electronic and optical properties of group III-V semiconductors: Arsenides and Antimonides
Investigating the structural, electronic, and optical properties of zinc-blende III-V semiconductors, particularly arsenides, and antimonides, which are crucial for optoelectronic devices such as transistors, infrared detectors, and quantum technologies due to their wide range of direct bandgaps. In this work, we have employed a first-principles approach integrating G0W0 with the HSE06 hybrid functional and spin–orbit coupling (SOC) to study their fundamental properties. Traditional Density Functional Theory (DFT) methods, particularly those using Generalized Gradient Approximation (GGA) PBE functionals, tend to underestimate bandgaps, leading to discrepancies with experimental results. To address this, our study corrects the bandgap underestimation and refines the calculation of optical constants, including the dielectric function, refractive index, extinction coefficient, and absorption coefficient. Moreover, the optimized lattice constants and electronic properties derived from our computational model strongly correlate with experimental data, demonstrating the model’s reliability in predicting material properties. The findings suggest that our methods can be applied to arsenides and antimonides, offering a pathway to designing materials with optoelectronic properties involving III-V compounds and their complex heterostructures for advanced device applications.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational Materials Science
工程技术-材料科学:综合
CiteScore
6.50
自引率
6.10%
发文量
665
审稿时长
26 days
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


