Krzysztof Kotlarz, Magda Mielczarek, Przemysław Biecek, Bernt Guldbrandtsen, Joanna Szyda
{"title":"利用基于人工智能的自动编码器方法,探索序列上下文对利用全基因组测序数据进行 SNP 基因型调用时出现错误的影响。","authors":"Krzysztof Kotlarz, Magda Mielczarek, Przemysław Biecek, Bernt Guldbrandtsen, Joanna Szyda","doi":"10.1093/nargab/lqae131","DOIUrl":null,"url":null,"abstract":"<p><p>A critical step in the analysis of whole genome sequencing data is variant calling. Despite its importance, variant calling is prone to errors. Our study investigated the association between incorrect single nucleotide polymorphism (SNP) calls and variant quality metrics and nucleotide context. In our study, incorrect SNPs were defined in 20 Holstein-Friesian cows by comparing their SNPs genotypes identified by whole genome sequencing with the IlluminaNovaSeq6000 and the EuroGMD50K genotyping microarray. The dataset was divided into the correct SNP set (666 333 SNPs) and the incorrect SNP set (4 557 SNPs). The training dataset consisted of only the correct SNPs, while the test dataset contained a balanced mix of all the incorrectly and correctly called SNPs. An autoencoder was constructed to identify systematically incorrect SNPs that were marked as outliers by a one-class support vector machine and isolation forest algorithms. The results showed that 59.53% (±0.39%) of the incorrect SNPs had systematic patterns, with the remainder being random errors. The frequent occurrence of the CGC 3-mer was due to mislabelling a call for C. Incorrect T instead of A call was associated with the presence of T in the neighbouring downstream position. These errors may arise due to the fluorescence patterns of nucleotide labelling.</p>","PeriodicalId":33994,"journal":{"name":"NAR Genomics and Bioinformatics","volume":"6 3","pages":"lqae131"},"PeriodicalIF":2.8000,"publicationDate":"2024-09-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11420682/pdf/","citationCount":"0","resultStr":"{\"title\":\"Exploring the impact of sequence context on errors in SNP genotype calling with whole genome sequencing data using AI-based autoencoder approach.\",\"authors\":\"Krzysztof Kotlarz, Magda Mielczarek, Przemysław Biecek, Bernt Guldbrandtsen, Joanna Szyda\",\"doi\":\"10.1093/nargab/lqae131\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>A critical step in the analysis of whole genome sequencing data is variant calling. Despite its importance, variant calling is prone to errors. Our study investigated the association between incorrect single nucleotide polymorphism (SNP) calls and variant quality metrics and nucleotide context. In our study, incorrect SNPs were defined in 20 Holstein-Friesian cows by comparing their SNPs genotypes identified by whole genome sequencing with the IlluminaNovaSeq6000 and the EuroGMD50K genotyping microarray. The dataset was divided into the correct SNP set (666 333 SNPs) and the incorrect SNP set (4 557 SNPs). The training dataset consisted of only the correct SNPs, while the test dataset contained a balanced mix of all the incorrectly and correctly called SNPs. An autoencoder was constructed to identify systematically incorrect SNPs that were marked as outliers by a one-class support vector machine and isolation forest algorithms. The results showed that 59.53% (±0.39%) of the incorrect SNPs had systematic patterns, with the remainder being random errors. The frequent occurrence of the CGC 3-mer was due to mislabelling a call for C. Incorrect T instead of A call was associated with the presence of T in the neighbouring downstream position. These errors may arise due to the fluorescence patterns of nucleotide labelling.</p>\",\"PeriodicalId\":33994,\"journal\":{\"name\":\"NAR Genomics and Bioinformatics\",\"volume\":\"6 3\",\"pages\":\"lqae131\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-09-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11420682/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NAR Genomics and Bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/nargab/lqae131\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Genomics and Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/nargab/lqae131","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
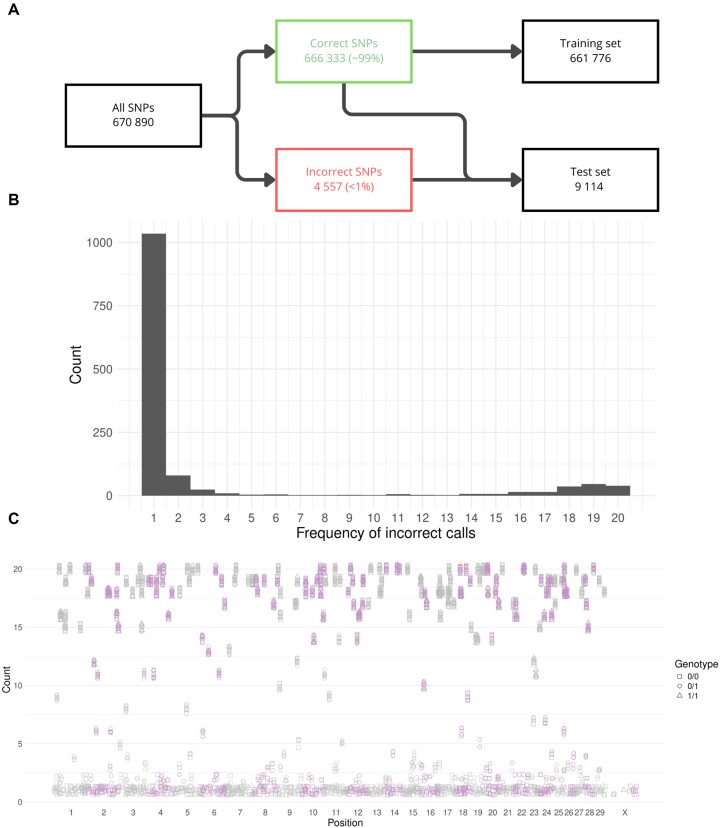
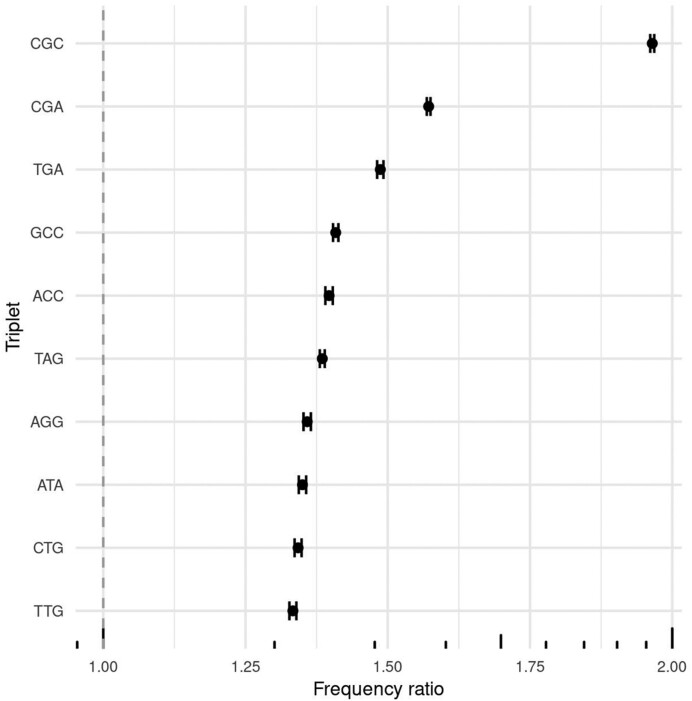
全基因组测序数据分析的一个关键步骤是变异体调用。尽管它很重要,但变异调用很容易出错。我们的研究调查了错误的单核苷酸多态性(SNP)调用与变异质量指标和核苷酸上下文之间的关联。在我们的研究中,通过比较使用 IlluminaNovaSeq6000 和 EuroGMD50K 基因分型芯片进行全基因组测序所确定的 SNP 基因型,对 20 头荷斯坦-弗里斯兰奶牛中不正确的 SNP 进行了定义。数据集分为正确 SNP 集(666 333 SNPs)和错误 SNP 集(4 557 SNPs)。训练数据集只包含正确的 SNP,而测试数据集则均衡地包含了所有错误和正确调用的 SNP。我们构建了一个自动编码器来识别系统错误的 SNP,这些 SNP 被单类支持向量机和隔离森林算法标记为异常值。结果显示,59.53%(±0.39%)的错误 SNP 具有系统模式,其余为随机误差。CGC 3-mer的频繁出现是由于错误标记了C的调用,而错误的T而不是A的调用与邻近下游位置存在T有关。这些错误可能是由于核苷酸标记的荧光模式造成的。
Exploring the impact of sequence context on errors in SNP genotype calling with whole genome sequencing data using AI-based autoencoder approach.
A critical step in the analysis of whole genome sequencing data is variant calling. Despite its importance, variant calling is prone to errors. Our study investigated the association between incorrect single nucleotide polymorphism (SNP) calls and variant quality metrics and nucleotide context. In our study, incorrect SNPs were defined in 20 Holstein-Friesian cows by comparing their SNPs genotypes identified by whole genome sequencing with the IlluminaNovaSeq6000 and the EuroGMD50K genotyping microarray. The dataset was divided into the correct SNP set (666 333 SNPs) and the incorrect SNP set (4 557 SNPs). The training dataset consisted of only the correct SNPs, while the test dataset contained a balanced mix of all the incorrectly and correctly called SNPs. An autoencoder was constructed to identify systematically incorrect SNPs that were marked as outliers by a one-class support vector machine and isolation forest algorithms. The results showed that 59.53% (±0.39%) of the incorrect SNPs had systematic patterns, with the remainder being random errors. The frequent occurrence of the CGC 3-mer was due to mislabelling a call for C. Incorrect T instead of A call was associated with the presence of T in the neighbouring downstream position. These errors may arise due to the fluorescence patterns of nucleotide labelling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


