通过深度学习解码邻近氨基酸对 ESI-MS 强度输出的影响。
IF 2.8
2区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
摘要
使用质谱(MS)进行肽级定量并非易事,因为理化特性会影响响应和可检测性。特定的氨基酸(AA)序列会影响这些特性,但人们对序列和强度输出之间的联系仍然知之甚少。在这项工作中,我们探索了氨基酸对的组合(即二聚体图案),以确定局部氨基酸环境与 MS1 强度之间的潜在关系。为此,我们建立了一个深度学习(DL)模型,该模型由带有注意机制的编码器-解码器组成。注意力机制可以识别最相关的主题。我们持续观察到了一些特定的模式,其中一个笨重/芳香和疏水 AA 之后是一个阳离子 AA,以及连续的笨重/芳香和疏水 AA 对于预测 MS1 强度非常重要。将注意力权重与平均 MS1 强度相关联发现,一些重要的主题(尤其是含有 Trp、His 和 Cys 的主题)与低响应肽相关联,而含有 Lys 和大多数笨重疏水 AA 的主题往往与高响应肽相关联。此外,Asn-Gly 与低响应相关。该模型预测 MS1 反应的平均百分比误差约为 11%,皮尔逊相关系数约为 0.68。虽然与早期工作中的单一 AA 表示相比,肽序列的二聚体表示并没有提高预测能力,但这项工作为更好地理解 MS 分析中的肽响应提供了宝贵的见解。意义:质谱本身并不是定量分析,化合物的响应不仅取决于其浓度,还取决于分子组成。对于基于质谱的肽段分析,如自下而上的蛋白质组学,这直接意味着响应不能直接用于定量单个肽段。此外,人们对响应与单个肽段的氨基酸序列之间的关系仍然知之甚少。在此,我们使用基于具有注意机制的递归神经网络的深度学习模型,通过分析等摩尔肽池中总共近 20 万个独特的肽段,研究了肽段中二聚体图案的存在如何影响 MS1 响应。我们不仅确定了某些二聚体类别和对响应有重大影响的特定二聚体,而且该模型还能在独立测试子集中以较低的误差率预测肽段强度。这些发现不仅提高了我们对肽序列与响应之间联系的理解,而且突出了利用深度学习开发绝对、无标记肽定量方法的潜力。本文章由计算机程序翻译,如有差异,请以英文原文为准。
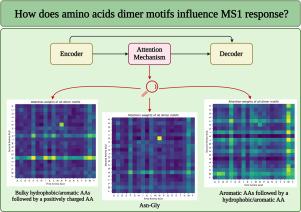
Decoding the impact of neighboring amino acids on ESI-MS intensity output through deep learning
Peptide-level quantification using mass spectrometry (MS) is no trivial task as the physicochemical properties affect both response and detectability. The specific amino acid (AA) sequence affects these properties, however the connection between sequence and intensity output remains poorly understood. In this work, we explore combinations of amino acid pairs (i.e., dimer motifs) to determine a potential relationship between the local amino acid environment and MS1 intensity. For this purpose, a deep learning (DL) model, consisting of an encoder-decoder with an attention mechanism, was built. The attention mechanism allowed to identify the most relevant motifs. Specific patterns were consistently observed where a bulky/aromatic and hydrophobic AA followed by a cationic AA as well as consecutive bulky/aromatic and hydrophobic AAs were found important for the prediction of the MS1 intensity. Correlating attention weights to mean MS1 intensities revealed that some important motifs, particularly containing Trp, His, and Cys, were linked with low responding peptides whereas motifs containing Lys and most bulky hydrophobic AAs were often associated with high responding peptides. Moreover, Asn-Gly was associated with low response. The model predicts MS1 response with a mean average percentage error of ∼11 % and a Pearson correlation coefficient of ∼0.64. While dimer representation of peptide sequences did not improve predictive capacity compared to single AA representation in earlier work, this work adds valuable insight for a better understanding of peptide response in MS analysis.
Significance
Mass spectrometry is not inherently quantitative, and the response of a compound relies not only on its concentration but also on the molecular composition. For mass spectrometry-based analysis of peptides, such as in bottom-up proteomics, this directly implies that the response cannot be used directly to quantify individual peptides. Moreover, the dependency of the response on the amino acid sequence of individual peptides remains poorly understood. Using a deep learning model based on a recurrent neural network with an attention mechanism, we here investigate how the presence of dimer motifs within a peptide affects the MS1 response through the analysis of intended equimolar peptide pools comprising almost 200,000 unique peptides in total. Not only do we identify certain dimer classes and specific dimers that substantially affect the MS1 response, but the model is also able to predict peptide intensity with low error rates within the independent test subset. The findings not only improve our understanding of the link between sequence and response for peptides but also highlight the potential of utilizing deep learning for developing methods allowing for absolute, label-free peptide quantification.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of proteomics
生物-生化研究方法
CiteScore
7.10
自引率
3.00%
发文量
227
审稿时长
73 days
期刊介绍:
Journal of Proteomics is aimed at protein scientists and analytical chemists in the field of proteomics, biomarker discovery, protein analytics, plant proteomics, microbial and animal proteomics, human studies, tissue imaging by mass spectrometry, non-conventional and non-model organism proteomics, and protein bioinformatics. The journal welcomes papers in new and upcoming areas such as metabolomics, genomics, systems biology, toxicogenomics, pharmacoproteomics.
Journal of Proteomics unifies both fundamental scientists and clinicians, and includes translational research. Suggestions for reviews, webinars and thematic issues are welcome.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


