Wfold:利用深度学习预测 RNA 二级结构的新方法。
IF 7
2区 医学
Q1 BIOLOGY
引用次数: 0
摘要
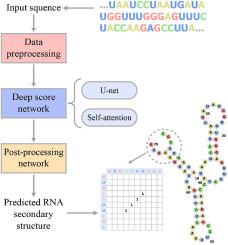
对 RNA 二级结构的精确估计有可能揭示非编码 RNA 在调节细胞活动中的各种作用。然而,传统的 RNA 二级结构预测方法主要依赖于通过自由能最小化的热动力模型,这是一个需要大量先验知识的费力过程。在此,建议使用基于端到端深度学习的方法 Wfold 进行 RNA 二级结构预测。Wfold 直接根据注释数据和碱基配对标准进行训练。它利用 RNA 序列的图像式表示,结合了变压器编码器的增强型 U-net 可以有效地处理这些图像。Wfold 将自我注意机制挖掘长程信息的优势与 U-net 收集局部信息的能力相结合,最终提高了 RNA 二级结构预测的准确性。我们使用族内和跨族的 RNA 数据集比较了 Wfold 的性能。当在不同的 RNA 家族上进行训练和评估时,它取得了与传统方法相似的性能,但在家族内数据集上的性能却大大超过了最先进的方法。此外,Wfold 还能可靠地预测假结点。研究结果表明,Wfold 可用于改进序列比对、功能注释和 RNA 结构建模。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Wfold: A new method for predicting RNA secondary structure with deep learning
Precise estimations of RNA secondary structures have the potential to reveal the various roles that non-coding RNAs play in regulating cellular activity. However, the mainstay of traditional RNA secondary structure prediction methods relies on thermos-dynamic models via free energy minimization, a laborious process that requires a lot of prior knowledge. Here, RNA secondary structure prediction using Wfold, an end-to-end deep learning-based approach, is suggested. Wfold is trained directly on annotated data and base-pairing criteria. It makes use of an image-like representation of RNA sequences, which an enhanced U-net incorporated with a transformer encoder can process effectively. Wfold eventually increases the accuracy of RNA secondary structure prediction by combining the benefits of self-attention mechanism's mining of long-range information with U-net's ability to gather local information. We compare Wfold's performance using RNA datasets that are within and across families. When trained and evaluated on different RNA families, it achieves a similar performance as the traditional methods, but dramatically outperforms the state-of-the-art methods on within-family datasets. Moreover, Wfold can also reliably forecast pseudoknots. The findings imply that Wfold may be useful for improving sequence alignment, functional annotations, and RNA structure modeling.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computers in biology and medicine
工程技术-工程:生物医学
CiteScore
11.70
自引率
10.40%
发文量
1086
审稿时长
74 days
期刊介绍:
Computers in Biology and Medicine is an international forum for sharing groundbreaking advancements in the use of computers in bioscience and medicine. This journal serves as a medium for communicating essential research, instruction, ideas, and information regarding the rapidly evolving field of computer applications in these domains. By encouraging the exchange of knowledge, we aim to facilitate progress and innovation in the utilization of computers in biology and medicine.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


