{"title":"通过结构感知深度学习预测 RNA 序列结构的可能性。","authors":"You Zhou, Giulia Pedrielli, Fei Zhang, Teresa Wu","doi":"10.1186/s12859-024-05916-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The active functionalities of RNA are recognized to be heavily dependent on the structure and sequence. Therefore, a model that can accurately evaluate a design by giving RNA sequence-structure pairs would be a valuable tool for many researchers. Machine learning methods have been explored to develop such tools, showing promising results. However, two key issues remain. Firstly, the performance of machine learning models is affected by the features used to characterize RNA. Currently, there is no consensus on which features are the most effective for characterizing RNA sequence-structure pairs. Secondly, most existing machine learning methods extract features describing entire RNA molecule. We argue that it is essential to define additional features that characterize nucleotides and specific sections of RNA structure to enhance the overall efficacy of the RNA design process.</p><p><strong>Results: </strong>We develop two deep learning models for evaluating RNA sequence-secondary structure pairs. The first model, NU-ResNet, uses a convolutional neural network architecture that solves the aforementioned problems by explicitly encoding RNA sequence-structure information into a 3D matrix. Building upon NU-ResNet, our second model, NUMO-ResNet, incorporates additional information derived from the characterizations of RNA, specifically the 2D folding motifs. In this work, we introduce an automated method to extract these motifs based on fundamental secondary structure descriptions. We evaluate the performance of both models on an independent testing dataset. Our proposed models outperform the models from literatures in this independent testing dataset. To assess the robustness of our models, we conduct 10-fold cross validation. To evaluate the generalization ability of NU-ResNet and NUMO-ResNet across different RNA families, we train and test our proposed models in different RNA families. Our proposed models show superior performance compared to the models from literatures when being tested across different independent RNA families.</p><p><strong>Conclusions: </strong>In this study, we propose two deep learning models, NU-ResNet and NUMO-ResNet, to evaluate RNA sequence-secondary structure pairs. These two models expand the field of data-driven approaches for learning RNA. Furthermore, these two models provide the new method to encode RNA sequence-secondary structure pairs.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"25 1","pages":"316"},"PeriodicalIF":3.3000,"publicationDate":"2024-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11443715/pdf/","citationCount":"0","resultStr":"{\"title\":\"Predicting RNA sequence-structure likelihood via structure-aware deep learning.\",\"authors\":\"You Zhou, Giulia Pedrielli, Fei Zhang, Teresa Wu\",\"doi\":\"10.1186/s12859-024-05916-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The active functionalities of RNA are recognized to be heavily dependent on the structure and sequence. Therefore, a model that can accurately evaluate a design by giving RNA sequence-structure pairs would be a valuable tool for many researchers. Machine learning methods have been explored to develop such tools, showing promising results. However, two key issues remain. Firstly, the performance of machine learning models is affected by the features used to characterize RNA. Currently, there is no consensus on which features are the most effective for characterizing RNA sequence-structure pairs. Secondly, most existing machine learning methods extract features describing entire RNA molecule. We argue that it is essential to define additional features that characterize nucleotides and specific sections of RNA structure to enhance the overall efficacy of the RNA design process.</p><p><strong>Results: </strong>We develop two deep learning models for evaluating RNA sequence-secondary structure pairs. The first model, NU-ResNet, uses a convolutional neural network architecture that solves the aforementioned problems by explicitly encoding RNA sequence-structure information into a 3D matrix. Building upon NU-ResNet, our second model, NUMO-ResNet, incorporates additional information derived from the characterizations of RNA, specifically the 2D folding motifs. In this work, we introduce an automated method to extract these motifs based on fundamental secondary structure descriptions. We evaluate the performance of both models on an independent testing dataset. Our proposed models outperform the models from literatures in this independent testing dataset. To assess the robustness of our models, we conduct 10-fold cross validation. To evaluate the generalization ability of NU-ResNet and NUMO-ResNet across different RNA families, we train and test our proposed models in different RNA families. Our proposed models show superior performance compared to the models from literatures when being tested across different independent RNA families.</p><p><strong>Conclusions: </strong>In this study, we propose two deep learning models, NU-ResNet and NUMO-ResNet, to evaluate RNA sequence-secondary structure pairs. These two models expand the field of data-driven approaches for learning RNA. Furthermore, these two models provide the new method to encode RNA sequence-secondary structure pairs.</p>\",\"PeriodicalId\":8958,\"journal\":{\"name\":\"BMC Bioinformatics\",\"volume\":\"25 1\",\"pages\":\"316\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-09-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11443715/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12859-024-05916-1\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-024-05916-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Predicting RNA sequence-structure likelihood via structure-aware deep learning.
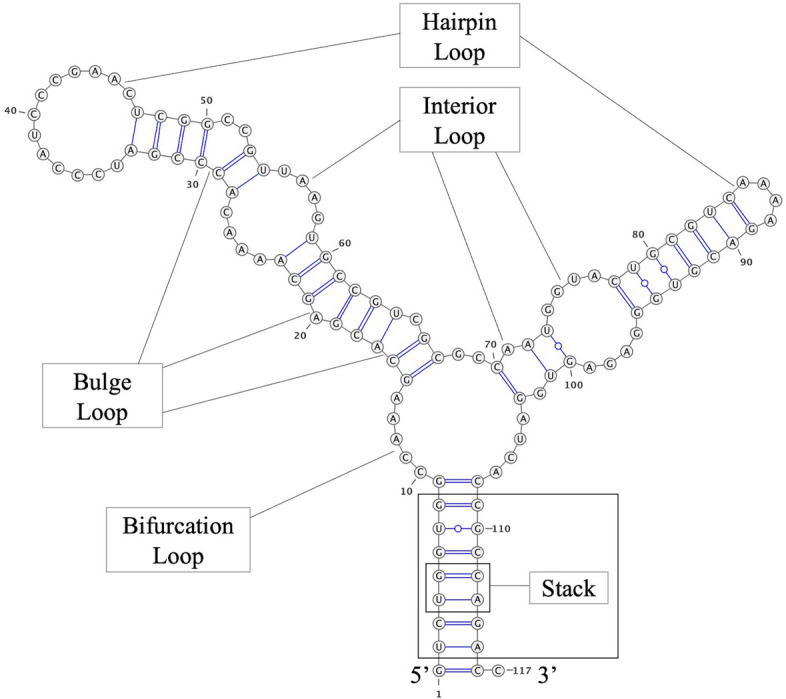
Background: The active functionalities of RNA are recognized to be heavily dependent on the structure and sequence. Therefore, a model that can accurately evaluate a design by giving RNA sequence-structure pairs would be a valuable tool for many researchers. Machine learning methods have been explored to develop such tools, showing promising results. However, two key issues remain. Firstly, the performance of machine learning models is affected by the features used to characterize RNA. Currently, there is no consensus on which features are the most effective for characterizing RNA sequence-structure pairs. Secondly, most existing machine learning methods extract features describing entire RNA molecule. We argue that it is essential to define additional features that characterize nucleotides and specific sections of RNA structure to enhance the overall efficacy of the RNA design process.
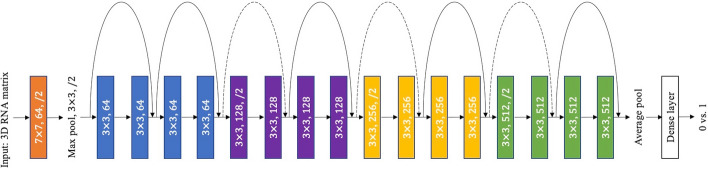
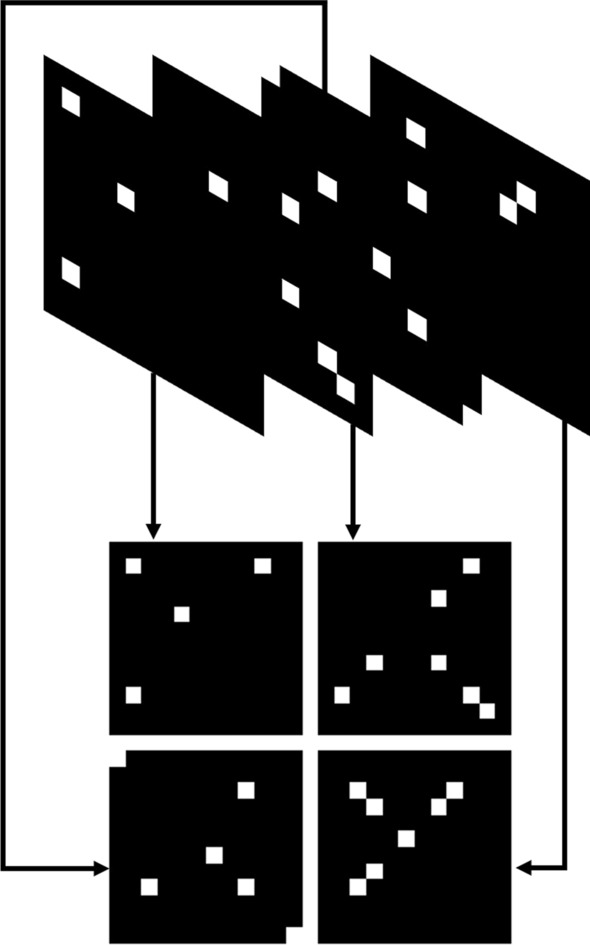
Results: We develop two deep learning models for evaluating RNA sequence-secondary structure pairs. The first model, NU-ResNet, uses a convolutional neural network architecture that solves the aforementioned problems by explicitly encoding RNA sequence-structure information into a 3D matrix. Building upon NU-ResNet, our second model, NUMO-ResNet, incorporates additional information derived from the characterizations of RNA, specifically the 2D folding motifs. In this work, we introduce an automated method to extract these motifs based on fundamental secondary structure descriptions. We evaluate the performance of both models on an independent testing dataset. Our proposed models outperform the models from literatures in this independent testing dataset. To assess the robustness of our models, we conduct 10-fold cross validation. To evaluate the generalization ability of NU-ResNet and NUMO-ResNet across different RNA families, we train and test our proposed models in different RNA families. Our proposed models show superior performance compared to the models from literatures when being tested across different independent RNA families.
Conclusions: In this study, we propose two deep learning models, NU-ResNet and NUMO-ResNet, to evaluate RNA sequence-secondary structure pairs. These two models expand the field of data-driven approaches for learning RNA. Furthermore, these two models provide the new method to encode RNA sequence-secondary structure pairs.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


