6-hydroxymethyl-7,8-dihydroptein pyrophosphokinase 的双底物抑制剂:细胞渗透性。
IF 2.5
4区 医学
Q3 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
6-羟甲基-7,8-二氢蝶呤焦磷激酶(HPPK)是叶酸生物合成途径中的一种关键酶。它催化 ATP 向 6-羟甲基-7,8-二氢蝶呤(HP)的焦磷酸转移。HPPK 在微生物中必不可少,但在哺乳动物中却不存在。然而,现有的抗生素都不是以它为靶标。因此,这种酶是开发新型抗菌剂的一个有吸引力的靶点。丰富的结构和机理信息为基于结构设计 HPPK 抑制剂提供了坚实的基础。我们的双底物抑制剂最初是通过 2、3 或 4 个磷酸基团(HPnA,n = 2、3 或 4)将 6-羟甲基蝶呤与腺苷连接在一起而产生的,其中 HP4A 表现出最高的结合亲和力(Kd = 0.47 ± 0.04 μM)。根据 HPPK 与 HP4A 复合物的高分辨率结构进行了进一步的开发。用哌啶连接的硫醚取代磷酸桥消除了桥上的多个负电荷。用 7,7-二甲基-7,8-二氢蝶呤取代蝶呤分子提高了结合亲和力。在哌啶环上添加羧基并氧化硫醚可进一步提高药效,从而产生类似药物的 HPPK 抑制剂(Kd = 0.047 ± 0.007 μM)。不过,这些抑制剂都不具有细菌细胞渗透性。这很可能是由于细菌中缺乏活性叶酸转运体。我们用 7-deazagaunine 分子取代了蝶呤分子,得到了一种新型双基质抑制剂(HP-101),它对革兰氏阳性细菌具有可观察到的细胞渗透性。在此,我们报告了 HP-101 的体外活性及其与 HPPK 复合物的结构,为基于结构的进一步开发提供了框架。本文章由计算机程序翻译,如有差异,请以英文原文为准。
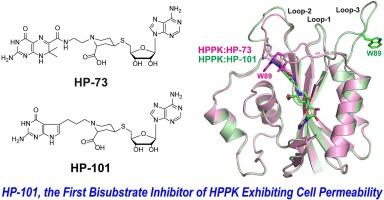
Bisubstrate inhibitors of 6-hydroxymethyl-7,8-dihydroptein pyrophosphokinase: Toward cell permeability
6-Hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK) is a key enzyme in the folate biosynthesis pathway. It catalyzes the pyrophosphoryl transfer from ATP to 6-hydroxymethyl-7,8-dihydropterin (HP). HPPK is essential for microorganisms but is absent in mammals. Yet, it is not the target of any existing antibiotics. Hence, this enzyme is an attractive target for developing novel antimicrobial agents. A wealth of structural and mechanistic information has provided solid basis for structure-based design of HPPK inhibitors. Our bisubstrate inhibitors were initially created by linking 6-hydroxymethylpterin to adenosine through 2, 3, or 4 phosphate groups (HPnA, n = 2, 3, or 4), among which HP4A exhibited the highest binding affinity (Kd = 0.47 ± 0.04 μM). Further development was carried out based on high-resolution structures of HPPK in complex with HP4A. Replacing the phosphate bridge with a piperidine linked thioether eliminated multiple negative charges of the bridge. Substituting the pterin moiety with 7,7-dimethyl-7,8-dihydropterin improved the binding affinity. Arming the piperidine ring with a carboxyl group and oxidizing the thioether further enhanced the potency, resulting in a druglike inhibitor of HPPK (Kd = 0.047 ± 0.007 μM). None of these inhibitors, however, exhibits bacterial cell permeability. It is most likely due to the lack of active folate transporters in bacteria. Replacing the pterin moiety with a 7-deazagaunine moiety, we have obtained a novel bisubstrate inhibitor (HP-101) showing observable cell permeability toward a Gram-positive bacterium. Here, we report the in vitro activity of HP-101 and its structure in complex with HPPK, providing a framework for structure-based further development.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
5.70
自引率
3.70%
发文量
463
审稿时长
27 days
期刊介绍:
Bioorganic & Medicinal Chemistry Letters presents preliminary experimental or theoretical research results of outstanding significance and timeliness on all aspects of science at the interface of chemistry and biology and on major advances in drug design and development. The journal publishes articles in the form of communications reporting experimental or theoretical results of special interest, and strives to provide maximum dissemination to a large, international audience.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


