In2O3(011) 表面预吸附的 H2 与 O2 反应的 DFT 模拟
IF 2.8
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
本文首次使用 DFT 方法来模拟氢与吸附在 In2O3(011) 缺陷表面上的氧分子的反应。该反应对 In2O3 传感器的氢气检测至关重要。活化能的计算采用了两种机制,涉及吸附水分子和 In2O3(011) 表面羟基的形成。一个羟基是由于羟基与表面金属原子成键而形成的,另一个是由于氢与晶格中的氧成键而形成的。这两个反应的特点都是存在势垒和放热。通过爬升图像推移弹性带法计算出这两个反应的活化能分别为 0.99 eV 和 0.98 eV。计算结果与现有实验数据进行了比较。计算结果还表明,表面中性氧空位的存在导致费米级以下空位态的形成,电子密度集中在四倍配位的铟原子上。本文章由计算机程序翻译,如有差异,请以英文原文为准。
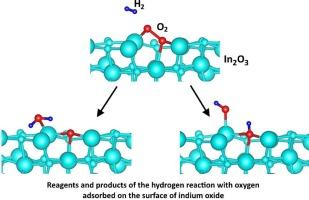
DFT modeling of reaction of H2 with O2 pre-adsorbed on In2O3(011) surface
The DFT approach is used for the first time to model the reaction of hydrogen with oxygen molecule adsorbed on the defective surface of In2O3(011). This reaction is crucial for hydrogen detection by In2O3 sensor. The activation energies are calculated using two mechanisms involving the formation of both an adsorbed water molecule and hydroxyl groups on the In2O3(011) surface. One hydroxyl group is formed due to the bonding of OH with the surface metal atom, and the other due to the bonding of hydrogen with oxygen of the lattice. Both reactions are characterized by the presence of a potential barrier and are exothermic. The activation energies of the two reactions are calculated by the climbing-image nudged elastic band method to be 0.99 eV and 0.98 eV. The results of the calculations are compared with available experimental data. It is also shown that the presence of a surface neutral oxygen vacancy leads to the formation of a vacancy state below the Fermi level, and the electron density is concentrated on the fourfold coordinated indium atom.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Chemical Physics Letters
化学-物理:原子、分子和化学物理
CiteScore
5.70
自引率
3.60%
发文量
798
审稿时长
33 days
期刊介绍:
Chemical Physics Letters has an open access mirror journal, Chemical Physics Letters: X, sharing the same aims and scope, editorial team, submission system and rigorous peer review.
Chemical Physics Letters publishes brief reports on molecules, interfaces, condensed phases, nanomaterials and nanostructures, polymers, biomolecular systems, and energy conversion and storage.
Criteria for publication are quality, urgency and impact. Further, experimental results reported in the journal have direct relevance for theory, and theoretical developments or non-routine computations relate directly to experiment. Manuscripts must satisfy these criteria and should not be minor extensions of previous work.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


