金、铜、MoO2 和 MoS2 二维纳米粒子吸附 CO 的比较研究
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
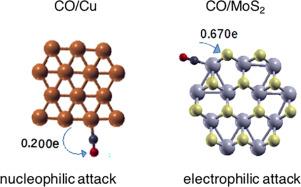
本研究的重点是比较分析由金、铜、二氧化硅和二氧化硅制成的含有 10、12 和 14 个图案的三角形、不规则六边形和八边形二维纳米粒子的电子特性。研究采用密度泛函理论进行。形成能量和振动频率表明,二维纳米结构构型可以作为稳定结构存在。配位数低于中心原子的边缘原子是吸附 CO 的首选位置。利用 Fukui 函数计算出的轨道加权双重描述符,可以在金和铜纳米粒子中识别出大部分亲核攻击位点,而基于 MoO2 和 MoS2 的纳米粒子则呈现出几乎与亲核位点一样多的亲电位点。纳米结构与 CO 分子之间转移的电荷以及投影状态密度的重新分布被用来评估界面键的强度以及键合中涉及的基本相互作用的性质。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Comparative study of CO adsorption on Au, Cu, MoO2 and MoS2 2D Nanoparticles
This study focuses on a comparative analysis of the electronic properties of triangular, irregular hexagonal, and octagonal 2D nanoparticles containing 10, 12, and 14 motifs, made of Au, Cu, MoO2, and MoS2. The investigation was carried out using density functional theory. The formation energies and vibrational frequencies demonstrate that the 2D nanostructure configurations can exist as stable structures. Edge atoms with lower coordination numbers than central atoms, are the preferred sites for CO adsorption. Using orbital-weighting dual descriptors calculated from Fukui functions enabled the identification of a majority nucleophilic attack sites in Au and Cu nanoparticles, while MoO2 and MoS2-based nanoparticles present almost as many electrophilic sites as nucleophilic sites. The charge transferred between the nanostructures and the CO molecule and the redistribution of the projected density of states were used to assess the strength of interfacial bonds and the nature of the fundamental interaction involved in the bonding.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


