Ryan J. Daniels, Britta S. Meyer, Marco Giulio, Silvia G. Signorini, Nicoletta Riccardi, Camilla Della Torre, Alexandra A.-T. Weber
{"title":"为自然种群表观基因组学的样本池设定基准","authors":"Ryan J. Daniels, Britta S. Meyer, Marco Giulio, Silvia G. Signorini, Nicoletta Riccardi, Camilla Della Torre, Alexandra A.-T. Weber","doi":"10.1111/1755-0998.14021","DOIUrl":null,"url":null,"abstract":"<p>DNA methylation (DNAm) is a mechanism for rapid acclimation to environmental conditions. In natural systems, small effect sizes relative to noise necessitates large sampling efforts to detect differences. Large numbers of individually sequenced libraries are costly. Pooling DNA prior to library preparation may be an efficient way to reduce costs and increase sample size, yet there are to date no recommendations in ecological epigenetics research. We test whether pooled and individual libraries yield comparable DNAm signals in a natural system exposed to different pollution levels by generating whole-epigenome data from two invasive molluscs (<i>Corbicula fluminea</i>, <i>Dreissena polymorpha</i>) collected from polluted and unpolluted localities (Italy). DNA of the same individuals were used for pooled and individual epigenomic libraries and sequenced with equivalent resources per individual. We found that pooling effectively captures similar genome-wide and global methylation signals as individual libraries, highlighting that pooled libraries are representative of the global population signal. However, pooled libraries yielded orders of magnitude more data than individual libraries, which was a consequence of higher coverage. We would therefore recommend aiming for a high initial coverage of individual libraries (15×) in future studies. Consequently, we detected many more differentially methylated regions (DMRs) with the pooled libraries and a significantly lower statistical power for regions from individual libraries. Computationally pooled data from the individual libraries produced fewer DMRs and the overlap with wet-lab pooled DMRs was relatively low. We discuss possible causes for discrepancies, list benefits and drawbacks of pooling, and provide recommendations for future epigenomic studies.</p>","PeriodicalId":211,"journal":{"name":"Molecular Ecology Resources","volume":"24 8","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/1755-0998.14021","citationCount":"0","resultStr":"{\"title\":\"Benchmarking sample pooling for epigenomics of natural populations\",\"authors\":\"Ryan J. Daniels, Britta S. Meyer, Marco Giulio, Silvia G. Signorini, Nicoletta Riccardi, Camilla Della Torre, Alexandra A.-T. Weber\",\"doi\":\"10.1111/1755-0998.14021\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>DNA methylation (DNAm) is a mechanism for rapid acclimation to environmental conditions. In natural systems, small effect sizes relative to noise necessitates large sampling efforts to detect differences. Large numbers of individually sequenced libraries are costly. Pooling DNA prior to library preparation may be an efficient way to reduce costs and increase sample size, yet there are to date no recommendations in ecological epigenetics research. We test whether pooled and individual libraries yield comparable DNAm signals in a natural system exposed to different pollution levels by generating whole-epigenome data from two invasive molluscs (<i>Corbicula fluminea</i>, <i>Dreissena polymorpha</i>) collected from polluted and unpolluted localities (Italy). DNA of the same individuals were used for pooled and individual epigenomic libraries and sequenced with equivalent resources per individual. We found that pooling effectively captures similar genome-wide and global methylation signals as individual libraries, highlighting that pooled libraries are representative of the global population signal. However, pooled libraries yielded orders of magnitude more data than individual libraries, which was a consequence of higher coverage. We would therefore recommend aiming for a high initial coverage of individual libraries (15×) in future studies. Consequently, we detected many more differentially methylated regions (DMRs) with the pooled libraries and a significantly lower statistical power for regions from individual libraries. Computationally pooled data from the individual libraries produced fewer DMRs and the overlap with wet-lab pooled DMRs was relatively low. We discuss possible causes for discrepancies, list benefits and drawbacks of pooling, and provide recommendations for future epigenomic studies.</p>\",\"PeriodicalId\":211,\"journal\":{\"name\":\"Molecular Ecology Resources\",\"volume\":\"24 8\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/1755-0998.14021\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Ecology Resources\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.14021\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Ecology Resources","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.14021","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
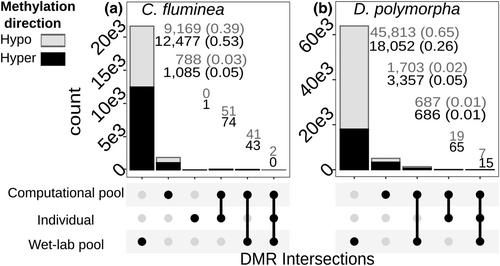
DNA 甲基化(DNAm)是快速适应环境条件的一种机制。在自然系统中,相对于噪声而言,效应大小较小,因此必须进行大量取样工作以检测差异。大量单独测序的文库成本高昂。在文库制备之前汇集 DNA 可能是降低成本和增加样本量的有效方法,但迄今为止还没有生态表观遗传学研究方面的建议。我们从意大利受污染和未受污染地区收集了两种入侵软体动物(Corbicula fluminea 和 Dreissena polymorpha)的全表观基因组数据,以检验在暴露于不同污染水平的自然系统中,集合文库和个体文库是否能产生相似的 DNAm 信号。相同个体的 DNA 被用于集合表观基因组文库和个体表观基因组文库,并在每个个体资源相当的情况下进行测序。我们发现,集合文库能有效捕获与单个文库相似的全基因组和全球甲基化信号,这表明集合文库能代表全球种群信号。不过,集合文库比单个文库获得的数据多出几个数量级,这是覆盖率较高的结果。因此,我们建议在今后的研究中将单个文库的初始覆盖率设定为高(15 倍)。因此,我们利用集合文库检测到了更多的差异甲基化区域(DMR),而单个文库检测到的区域的统计能力则明显较低。计算汇集的单个文库数据产生的 DMRs 较少,而且与湿实验室汇集的 DMRs 重叠率相对较低。我们讨论了造成差异的可能原因,列出了汇集的优点和缺点,并为未来的表观基因组研究提供了建议。
Benchmarking sample pooling for epigenomics of natural populations
DNA methylation (DNAm) is a mechanism for rapid acclimation to environmental conditions. In natural systems, small effect sizes relative to noise necessitates large sampling efforts to detect differences. Large numbers of individually sequenced libraries are costly. Pooling DNA prior to library preparation may be an efficient way to reduce costs and increase sample size, yet there are to date no recommendations in ecological epigenetics research. We test whether pooled and individual libraries yield comparable DNAm signals in a natural system exposed to different pollution levels by generating whole-epigenome data from two invasive molluscs (Corbicula fluminea, Dreissena polymorpha) collected from polluted and unpolluted localities (Italy). DNA of the same individuals were used for pooled and individual epigenomic libraries and sequenced with equivalent resources per individual. We found that pooling effectively captures similar genome-wide and global methylation signals as individual libraries, highlighting that pooled libraries are representative of the global population signal. However, pooled libraries yielded orders of magnitude more data than individual libraries, which was a consequence of higher coverage. We would therefore recommend aiming for a high initial coverage of individual libraries (15×) in future studies. Consequently, we detected many more differentially methylated regions (DMRs) with the pooled libraries and a significantly lower statistical power for regions from individual libraries. Computationally pooled data from the individual libraries produced fewer DMRs and the overlap with wet-lab pooled DMRs was relatively low. We discuss possible causes for discrepancies, list benefits and drawbacks of pooling, and provide recommendations for future epigenomic studies.
期刊介绍:
Molecular Ecology Resources promotes the creation of comprehensive resources for the scientific community, encompassing computer programs, statistical and molecular advancements, and a diverse array of molecular tools. Serving as a conduit for disseminating these resources, the journal targets a broad audience of researchers in the fields of evolution, ecology, and conservation. Articles in Molecular Ecology Resources are crafted to support investigations tackling significant questions within these disciplines.
In addition to original resource articles, Molecular Ecology Resources features Reviews, Opinions, and Comments relevant to the field. The journal also periodically releases Special Issues focusing on resource development within specific areas.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


