Jinxiang Liu , Yongqi Yuan , Sikai Wang , Jiaheng Wang , Shengli Liu
{"title":"从分子角度深入了解二氧化碳水合物促进剂的性能","authors":"Jinxiang Liu , Yongqi Yuan , Sikai Wang , Jiaheng Wang , Shengli Liu","doi":"10.1016/j.jmgm.2024.108868","DOIUrl":null,"url":null,"abstract":"<div><p>Hydrate-based CO<sub>2</sub> storage is a cost-effective and environmentally friendly approach to reduce carbon emission, and the addition of hydrate promoters has shown a promising avenue for enhancing CO<sub>2</sub> hydrate formation. In this work, the promotion mechanism and promotion performance of five different hydrate promoters (denoted as DIOX, CP, THF, THP, and CH) were investigated and compared by first-principles calculations and molecular dynamics simulations. The results show that the hydrate promoters prefer to singly occupy 5<sup>12</sup>6<sup>4</sup> cages of the sII hydrate, and CO<sub>2</sub> molecules can singly occupy 5<sup>12</sup> cage or multiply occupy 5<sup>12</sup>6<sup>4</sup> cages. The cohesive energy density indicates that the optimum CO<sub>2</sub> storage capacity can reach up to ∼28 wt%. The stabilization effects of hydrate promoters on the hydrate stability should follow the order of CP > CH > DIOX > THF ≈ THP. The hydrate promoters can increase the water-water interactions, and the molecular diffusivity shows that the dynamic stability of the hydrates is THP ≈ CH > CP > DIOX > THF. Further, the hydrate promoters can accelerate the hydrate formation kinetics, which reduce the induction time and increase the nucleation and growth process.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"133 ","pages":"Article 108868"},"PeriodicalIF":2.7000,"publicationDate":"2024-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Molecular insights into the performance of promoters for carbon dioxide hydrate\",\"authors\":\"Jinxiang Liu , Yongqi Yuan , Sikai Wang , Jiaheng Wang , Shengli Liu\",\"doi\":\"10.1016/j.jmgm.2024.108868\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Hydrate-based CO<sub>2</sub> storage is a cost-effective and environmentally friendly approach to reduce carbon emission, and the addition of hydrate promoters has shown a promising avenue for enhancing CO<sub>2</sub> hydrate formation. In this work, the promotion mechanism and promotion performance of five different hydrate promoters (denoted as DIOX, CP, THF, THP, and CH) were investigated and compared by first-principles calculations and molecular dynamics simulations. The results show that the hydrate promoters prefer to singly occupy 5<sup>12</sup>6<sup>4</sup> cages of the sII hydrate, and CO<sub>2</sub> molecules can singly occupy 5<sup>12</sup> cage or multiply occupy 5<sup>12</sup>6<sup>4</sup> cages. The cohesive energy density indicates that the optimum CO<sub>2</sub> storage capacity can reach up to ∼28 wt%. The stabilization effects of hydrate promoters on the hydrate stability should follow the order of CP > CH > DIOX > THF ≈ THP. The hydrate promoters can increase the water-water interactions, and the molecular diffusivity shows that the dynamic stability of the hydrates is THP ≈ CH > CP > DIOX > THF. Further, the hydrate promoters can accelerate the hydrate formation kinetics, which reduce the induction time and increase the nucleation and growth process.</p></div>\",\"PeriodicalId\":16361,\"journal\":{\"name\":\"Journal of molecular graphics & modelling\",\"volume\":\"133 \",\"pages\":\"Article 108868\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular graphics & modelling\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1093326324001682\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001682","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Molecular insights into the performance of promoters for carbon dioxide hydrate
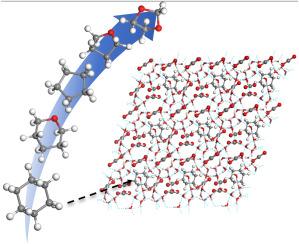
Hydrate-based CO2 storage is a cost-effective and environmentally friendly approach to reduce carbon emission, and the addition of hydrate promoters has shown a promising avenue for enhancing CO2 hydrate formation. In this work, the promotion mechanism and promotion performance of five different hydrate promoters (denoted as DIOX, CP, THF, THP, and CH) were investigated and compared by first-principles calculations and molecular dynamics simulations. The results show that the hydrate promoters prefer to singly occupy 51264 cages of the sII hydrate, and CO2 molecules can singly occupy 512 cage or multiply occupy 51264 cages. The cohesive energy density indicates that the optimum CO2 storage capacity can reach up to ∼28 wt%. The stabilization effects of hydrate promoters on the hydrate stability should follow the order of CP > CH > DIOX > THF ≈ THP. The hydrate promoters can increase the water-water interactions, and the molecular diffusivity shows that the dynamic stability of the hydrates is THP ≈ CH > CP > DIOX > THF. Further, the hydrate promoters can accelerate the hydrate formation kinetics, which reduce the induction time and increase the nucleation and growth process.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


