Alexander S. Ryzhako, Anna A. Tuma, Arseniy A. Otlyotov, Yury Minenkov
{"title":"电子结构理论方法、热力学和隐式溶解修正对有机碳酸盐构象和结合能的影响","authors":"Alexander S. Ryzhako, Anna A. Tuma, Arseniy A. Otlyotov, Yury Minenkov","doi":"10.1002/jcc.27471","DOIUrl":null,"url":null,"abstract":"<p>An impact of an electronic structure or force field method, gas-phase thermodynamic correction, and continuum solvation model on organic carbonate clusters (S)<sub><i>n</i></sub> conformational and binding energies is explored. None of the tested force field (GFN-FF, GAFF, MMFF94) and standard semiempirical methods (PM3, AM1, RM1, PM6, PM6-D3, PM6-D3H4, PM7) can reproduce reference RI-SCS-MP2 conformational energies. Tight-binding GFN<i>n</i>-xTB methods provide more realistic conformational energies which are accurate enough to discard the least stable conformers. The effect of thermodynamic correction is moderate and can be ignored if the gas phase conformational stability ranking is a goal. The influence of continuum solvation is stronger, especially if reinforced with the Gibbs free energy thermodynamic correction, and results in the reduced spread of conformational energies. The cluster formation binding energies strongly depend on a particular approach to vibrational thermochemistry with the difference between traditional harmonic and modified scaled rigid – harmonic oscillator approximations reaching 10 kcal mol<sup>−1</sup>.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"3004-3016"},"PeriodicalIF":3.4000,"publicationDate":"2024-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"An influence of electronic structure theory method, thermodynamic and implicit solvation corrections on the organic carbonates conformational and binding energies\",\"authors\":\"Alexander S. Ryzhako, Anna A. Tuma, Arseniy A. Otlyotov, Yury Minenkov\",\"doi\":\"10.1002/jcc.27471\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>An impact of an electronic structure or force field method, gas-phase thermodynamic correction, and continuum solvation model on organic carbonate clusters (S)<sub><i>n</i></sub> conformational and binding energies is explored. None of the tested force field (GFN-FF, GAFF, MMFF94) and standard semiempirical methods (PM3, AM1, RM1, PM6, PM6-D3, PM6-D3H4, PM7) can reproduce reference RI-SCS-MP2 conformational energies. Tight-binding GFN<i>n</i>-xTB methods provide more realistic conformational energies which are accurate enough to discard the least stable conformers. The effect of thermodynamic correction is moderate and can be ignored if the gas phase conformational stability ranking is a goal. The influence of continuum solvation is stronger, especially if reinforced with the Gibbs free energy thermodynamic correction, and results in the reduced spread of conformational energies. The cluster formation binding energies strongly depend on a particular approach to vibrational thermochemistry with the difference between traditional harmonic and modified scaled rigid – harmonic oscillator approximations reaching 10 kcal mol<sup>−1</sup>.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 32\",\"pages\":\"3004-3016\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-09-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27471\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27471","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
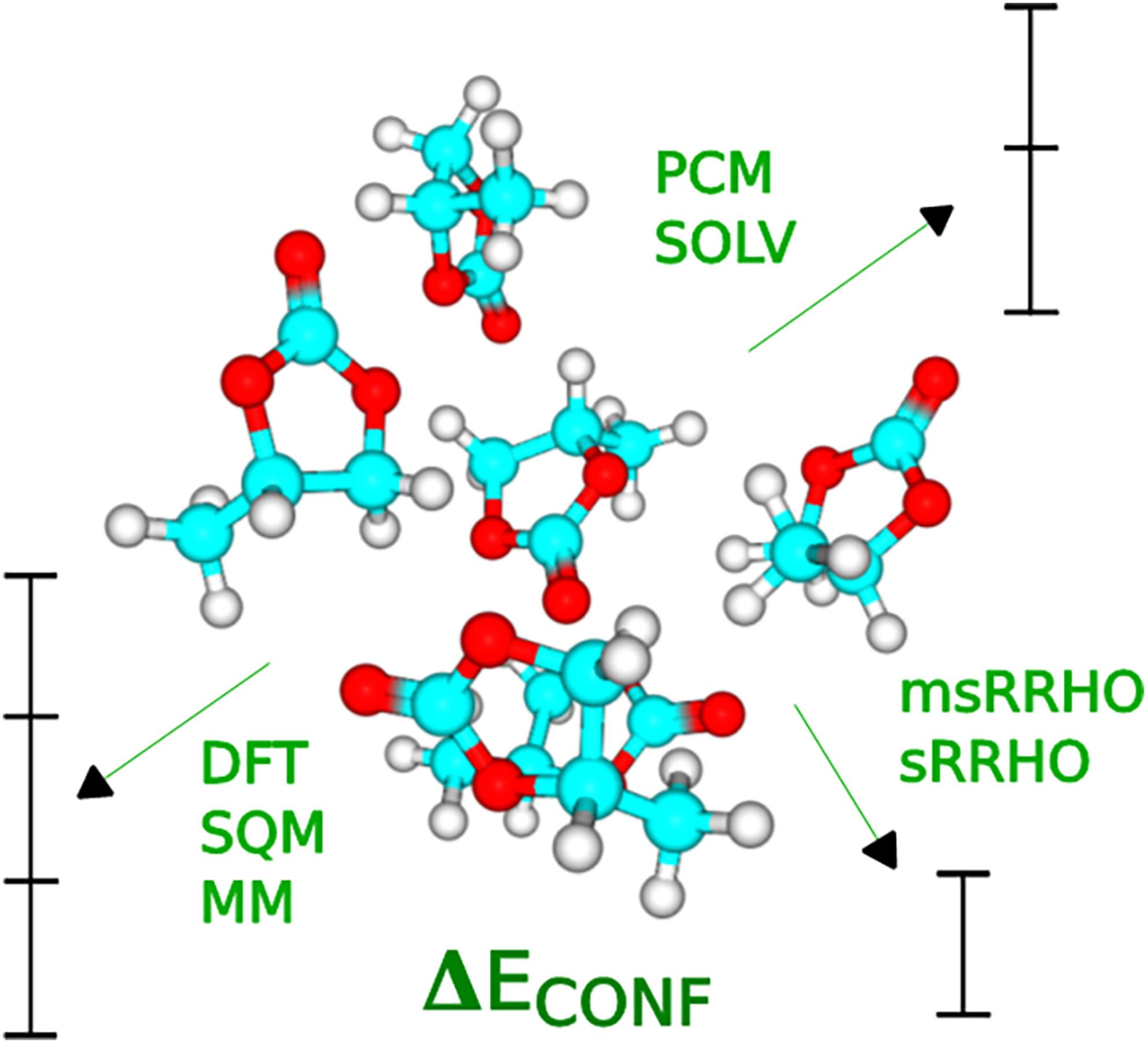
An influence of electronic structure theory method, thermodynamic and implicit solvation corrections on the organic carbonates conformational and binding energies
An impact of an electronic structure or force field method, gas-phase thermodynamic correction, and continuum solvation model on organic carbonate clusters (S)n conformational and binding energies is explored. None of the tested force field (GFN-FF, GAFF, MMFF94) and standard semiempirical methods (PM3, AM1, RM1, PM6, PM6-D3, PM6-D3H4, PM7) can reproduce reference RI-SCS-MP2 conformational energies. Tight-binding GFNn-xTB methods provide more realistic conformational energies which are accurate enough to discard the least stable conformers. The effect of thermodynamic correction is moderate and can be ignored if the gas phase conformational stability ranking is a goal. The influence of continuum solvation is stronger, especially if reinforced with the Gibbs free energy thermodynamic correction, and results in the reduced spread of conformational energies. The cluster formation binding energies strongly depend on a particular approach to vibrational thermochemistry with the difference between traditional harmonic and modified scaled rigid – harmonic oscillator approximations reaching 10 kcal mol−1.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


