Thilini Maheshika Herath, Bei Zhang, Dhimas Dwinandha and Manabu Fujii*,
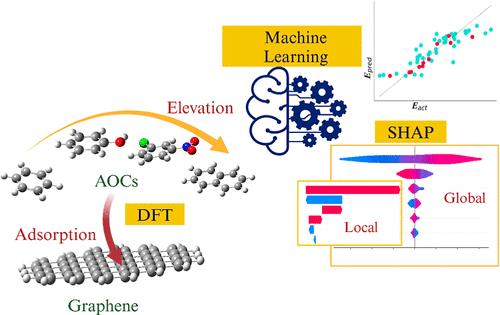
{"title":"通过量子化学计算与可解释机器学习相结合,阐明石墨烯材料对新兴芳香族有机污染物的吸附机理和特征","authors":"Thilini Maheshika Herath, Bei Zhang, Dhimas Dwinandha and Manabu Fujii*, ","doi":"10.1021/acsestwater.4c0021910.1021/acsestwater.4c00219","DOIUrl":null,"url":null,"abstract":"<p >As a complementary or alternative approach to experiments, theoretical computation of adsorption between carbon materials and emerging aromatic organic contaminants (AOCs) is increasingly important in elucidating adsorption mechanisms and characteristics, as well as their predictions. In this study, the adsorption energies between graphene and 112 AOCs were first analyzed by density functional theory (DFT-D). By the use of quantum molecular descriptors, different machine learning (ML) algorithms were developed. EXtreme gradient boosting exhibited the best performance among the four ML algorithms investigated, showing the lowest root-mean-square percentage error of 4.5% for the test data set. Accordingly, the interpretable ML technique (i.e., SHAP) assessed the importance and dependence of descriptors in the adsorption mechanisms of AOCs to graphene. The global interpretation confirmed that the molecular-volume-induced van der Waals interactions including π–π stacking are dominant, whereas the other interactions (e.g., induced hydrogen and electrostatic interactions) are comparably less significant in the adsorption of most AOCs on graphene. In contrast, using local interpretation, hydrogen bonds and induced dipole interactions with surrounding water were identified as important explanatory variables in the adsorption of AOCs containing carbonyl and sulfur functional groups. Therefore, the developed DFT-D-based ML models could be a reference model for theoretical and experimental studies.</p>","PeriodicalId":93847,"journal":{"name":"ACS ES&T water","volume":"4 9","pages":"3918–3930 3918–3930"},"PeriodicalIF":4.8000,"publicationDate":"2024-08-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Elucidating Adsorption Mechanisms and Characteristics of Emerging Aromatic Organic Contaminants to Graphene Material by Quantum Chemical Calculation Integrated with Interpretable Machine Learning\",\"authors\":\"Thilini Maheshika Herath, Bei Zhang, Dhimas Dwinandha and Manabu Fujii*, \",\"doi\":\"10.1021/acsestwater.4c0021910.1021/acsestwater.4c00219\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >As a complementary or alternative approach to experiments, theoretical computation of adsorption between carbon materials and emerging aromatic organic contaminants (AOCs) is increasingly important in elucidating adsorption mechanisms and characteristics, as well as their predictions. In this study, the adsorption energies between graphene and 112 AOCs were first analyzed by density functional theory (DFT-D). By the use of quantum molecular descriptors, different machine learning (ML) algorithms were developed. EXtreme gradient boosting exhibited the best performance among the four ML algorithms investigated, showing the lowest root-mean-square percentage error of 4.5% for the test data set. Accordingly, the interpretable ML technique (i.e., SHAP) assessed the importance and dependence of descriptors in the adsorption mechanisms of AOCs to graphene. The global interpretation confirmed that the molecular-volume-induced van der Waals interactions including π–π stacking are dominant, whereas the other interactions (e.g., induced hydrogen and electrostatic interactions) are comparably less significant in the adsorption of most AOCs on graphene. In contrast, using local interpretation, hydrogen bonds and induced dipole interactions with surrounding water were identified as important explanatory variables in the adsorption of AOCs containing carbonyl and sulfur functional groups. Therefore, the developed DFT-D-based ML models could be a reference model for theoretical and experimental studies.</p>\",\"PeriodicalId\":93847,\"journal\":{\"name\":\"ACS ES&T water\",\"volume\":\"4 9\",\"pages\":\"3918–3930 3918–3930\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2024-08-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS ES&T water\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acsestwater.4c00219\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ENVIRONMENTAL SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS ES&T water","FirstCategoryId":"1085","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsestwater.4c00219","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENVIRONMENTAL SCIENCES","Score":null,"Total":0}
引用次数: 0
摘要
作为实验的补充或替代方法,碳材料与新出现的芳香族有机污染物(AOC)之间的吸附理论计算在阐明吸附机理、特征及其预测方面越来越重要。本研究首先利用密度泛函理论(DFT-D)分析了石墨烯与 112 种芳香族有机污染物之间的吸附能。利用量子分子描述符,开发了不同的机器学习(ML)算法。在所研究的四种 ML 算法中,EXtreme gradient boosting 算法表现最佳,在测试数据集上显示出最低的均方根百分比误差(4.5%)。因此,可解释的 ML 技术(即 SHAP)评估了描述符在 AOC 对石墨烯吸附机制中的重要性和依赖性。全局解释证实,在大多数 AOCs 在石墨烯上的吸附过程中,分子体积诱导的范德华相互作用(包括 π-π 堆积)占主导地位,而其他相互作用(如诱导的氢相互作用和静电相互作用)的重要性则相对较低。相反,通过局部解释,氢键和与周围水的诱导偶极相互作用被认为是含羰基和硫官能团的 AOC 吸附过程中的重要解释变量。因此,所建立的基于 DFT-D 的 ML 模型可以作为理论和实验研究的参考模型。
Elucidating Adsorption Mechanisms and Characteristics of Emerging Aromatic Organic Contaminants to Graphene Material by Quantum Chemical Calculation Integrated with Interpretable Machine Learning
As a complementary or alternative approach to experiments, theoretical computation of adsorption between carbon materials and emerging aromatic organic contaminants (AOCs) is increasingly important in elucidating adsorption mechanisms and characteristics, as well as their predictions. In this study, the adsorption energies between graphene and 112 AOCs were first analyzed by density functional theory (DFT-D). By the use of quantum molecular descriptors, different machine learning (ML) algorithms were developed. EXtreme gradient boosting exhibited the best performance among the four ML algorithms investigated, showing the lowest root-mean-square percentage error of 4.5% for the test data set. Accordingly, the interpretable ML technique (i.e., SHAP) assessed the importance and dependence of descriptors in the adsorption mechanisms of AOCs to graphene. The global interpretation confirmed that the molecular-volume-induced van der Waals interactions including π–π stacking are dominant, whereas the other interactions (e.g., induced hydrogen and electrostatic interactions) are comparably less significant in the adsorption of most AOCs on graphene. In contrast, using local interpretation, hydrogen bonds and induced dipole interactions with surrounding water were identified as important explanatory variables in the adsorption of AOCs containing carbonyl and sulfur functional groups. Therefore, the developed DFT-D-based ML models could be a reference model for theoretical and experimental studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


