{"title":"三个中国家庭中与先天性蛛网膜畸形相关的FBN2缺失变异基因","authors":"Yu Sui, Yongping Lu, Meina Lin, Xinren Chen, Xiang Ni, Huan Li, Miao Jiang","doi":"10.1016/j.ymgmr.2024.101140","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>Congenital contractural arachnodactyly (CCA) is a rare autosomal dominant disorder caused by pathogenic variants of Fibrillin-2 (<em>FBN2</em>) gene. This study aimed to investigate the variants in three Chinese families with CCA.</p></div><div><h3>Methods</h3><p>Next-generation sequencing analysis and Sanger sequencing of exons 24–35 of <em>FBN2</em> (NM_001999.4) were performed on the three CCA pedigrees. The pathogenicity of the variants was assessed using ACMG criteria and predicted using an in-silico program.</p></div><div><h3>Results</h3><p>A novel heterozygous substitution (NM_001999.4: c.3230G > A; NP_001990.2 p. Cys1077Tyr) was identified in all patients from pedigree A, but not in healthy family members. The variant was found to be pathogenic. Additionally, in pedigree B (NM_001999.4: c.4222G > A; NP_001990.2: p.Asp1408Asn) and C (NM_001999.4: c.3170G > A; NP_001990.2: p.Gly1057Asp), and the previously reported variants were detected. Variants affecting cysteine residues may disrupt disulfide bridging, leading to a weakened microfibril scaffold, resulting in CCA phenotypes. High phenotypic heterogeneity was observed among different families, and there was little correlation between the genotype and phenotype.</p></div><div><h3>Conclusion</h3><p>This study describes three large families with CCA caused by missense variants in the <em>FBN2</em> gene. Phenotypic variations were observed among different pedigree groups, and further research is needed to investigate the underlying reasons for these variations.</p></div>","PeriodicalId":18814,"journal":{"name":"Molecular Genetics and Metabolism Reports","volume":"41 ","pages":"Article 101140"},"PeriodicalIF":1.8000,"publicationDate":"2024-09-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2214426924000934/pdfft?md5=65ae1d4fa45d800bf053f549131f8b0e&pid=1-s2.0-S2214426924000934-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Missense variants of FBN2 associated with congenital arachnodactyly in three Chinese families\",\"authors\":\"Yu Sui, Yongping Lu, Meina Lin, Xinren Chen, Xiang Ni, Huan Li, Miao Jiang\",\"doi\":\"10.1016/j.ymgmr.2024.101140\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Background</h3><p>Congenital contractural arachnodactyly (CCA) is a rare autosomal dominant disorder caused by pathogenic variants of Fibrillin-2 (<em>FBN2</em>) gene. This study aimed to investigate the variants in three Chinese families with CCA.</p></div><div><h3>Methods</h3><p>Next-generation sequencing analysis and Sanger sequencing of exons 24–35 of <em>FBN2</em> (NM_001999.4) were performed on the three CCA pedigrees. The pathogenicity of the variants was assessed using ACMG criteria and predicted using an in-silico program.</p></div><div><h3>Results</h3><p>A novel heterozygous substitution (NM_001999.4: c.3230G > A; NP_001990.2 p. Cys1077Tyr) was identified in all patients from pedigree A, but not in healthy family members. The variant was found to be pathogenic. Additionally, in pedigree B (NM_001999.4: c.4222G > A; NP_001990.2: p.Asp1408Asn) and C (NM_001999.4: c.3170G > A; NP_001990.2: p.Gly1057Asp), and the previously reported variants were detected. Variants affecting cysteine residues may disrupt disulfide bridging, leading to a weakened microfibril scaffold, resulting in CCA phenotypes. High phenotypic heterogeneity was observed among different families, and there was little correlation between the genotype and phenotype.</p></div><div><h3>Conclusion</h3><p>This study describes three large families with CCA caused by missense variants in the <em>FBN2</em> gene. Phenotypic variations were observed among different pedigree groups, and further research is needed to investigate the underlying reasons for these variations.</p></div>\",\"PeriodicalId\":18814,\"journal\":{\"name\":\"Molecular Genetics and Metabolism Reports\",\"volume\":\"41 \",\"pages\":\"Article 101140\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-09-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S2214426924000934/pdfft?md5=65ae1d4fa45d800bf053f549131f8b0e&pid=1-s2.0-S2214426924000934-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics and Metabolism Reports\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2214426924000934\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics and Metabolism Reports","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2214426924000934","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
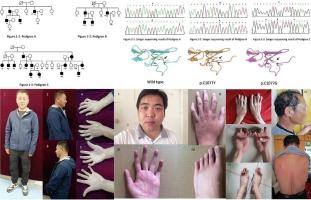
背景先天性挛缩性蛛网膜挛缩症(CCA)是一种罕见的常染色体显性遗传疾病,由Fibrillin-2(FBN2)基因的致病变异引起。本研究旨在调查三个中国 CCA 家系中的变体。方法对三个 CCA 家系的 FBN2(NM_001999.4)外显子 24-35 进行了下一代测序分析和 Sanger 测序。结果 在血统 A 的所有患者中发现了一个新的杂合子置换(NM_001999.4:c.3230G > A; NP_001990.2 p. Cys1077Tyr),但在健康的家庭成员中没有发现。发现该变异具有致病性。此外,在血统 B(NM_001999.4:c.4222G > A; NP_001990.2:p.Asp1408Asn)和 C(NM_001999.4:c.3170G > A; NP_001990.2:p.Gly1057Asp)中,也检测到了之前报告的变异。影响半胱氨酸残基的变异可能会破坏二硫桥,导致微纤维支架减弱,从而产生 CCA 表型。本研究描述了三个由 FBN2 基因错义变异引起的 CCA 家族。不同血统组之间存在表型差异,需要进一步研究这些差异的根本原因。
Missense variants of FBN2 associated with congenital arachnodactyly in three Chinese families
Background
Congenital contractural arachnodactyly (CCA) is a rare autosomal dominant disorder caused by pathogenic variants of Fibrillin-2 (FBN2) gene. This study aimed to investigate the variants in three Chinese families with CCA.
Methods
Next-generation sequencing analysis and Sanger sequencing of exons 24–35 of FBN2 (NM_001999.4) were performed on the three CCA pedigrees. The pathogenicity of the variants was assessed using ACMG criteria and predicted using an in-silico program.
Results
A novel heterozygous substitution (NM_001999.4: c.3230G > A; NP_001990.2 p. Cys1077Tyr) was identified in all patients from pedigree A, but not in healthy family members. The variant was found to be pathogenic. Additionally, in pedigree B (NM_001999.4: c.4222G > A; NP_001990.2: p.Asp1408Asn) and C (NM_001999.4: c.3170G > A; NP_001990.2: p.Gly1057Asp), and the previously reported variants were detected. Variants affecting cysteine residues may disrupt disulfide bridging, leading to a weakened microfibril scaffold, resulting in CCA phenotypes. High phenotypic heterogeneity was observed among different families, and there was little correlation between the genotype and phenotype.
Conclusion
This study describes three large families with CCA caused by missense variants in the FBN2 gene. Phenotypic variations were observed among different pedigree groups, and further research is needed to investigate the underlying reasons for these variations.
期刊介绍:
Molecular Genetics and Metabolism Reports is an open access journal that publishes molecular and metabolic reports describing investigations that use the tools of biochemistry and molecular biology for studies of normal and diseased states. In addition to original research articles, sequence reports, brief communication reports and letters to the editor are considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


