Tshele Mokhantso, Dean Sherry, Roland Worth, Ramesh Pandian, Ikechukwu Achilonu, Yasien Sayed
{"title":"铰链区插入和非活性位点突变对 HIV 蛋白酶-抑制剂相互作用的影响对比:从改变的瓣膜动力学中获得启示。","authors":"Tshele Mokhantso, Dean Sherry, Roland Worth, Ramesh Pandian, Ikechukwu Achilonu, Yasien Sayed","doi":"10.1016/j.jmgm.2024.108850","DOIUrl":null,"url":null,"abstract":"<div><p>HIV-1 protease (PR) enzyme is a viable antiretroviral drug target due to its crucial role in HIV maturation. Over many decades, the HIV-1 PR enzyme has exhibited mutations brought on by drug pressure and error-prone nature of HIV-1 reverse transcriptase. Non-active site mutations have played a pivotal role in drug resistance; however, their mechanism of action has not been fully elucidated. We investigated how non-active site mutations affect the conformational stability and drug binding ability of HIV-1 PR. In light of this, we studied a novel HIV-1 subtype C protease variant containing an insertion of valine (↑V) in the hinge region. We analysed the mutations in the presence and absence of ten background mutations. Molecular dynamics simulations revealed that both with and without the background mutations, the PR exhibited increased flexibility of hinge, flaps and fulcrum regions. This allowed the PR to adopt a wider flap conformation when in complex with several inhibitors. Additionally, the simulations revealed that the protease inhibitors (PIs) could not bring the mutated variant proteases into a stable, closed conformation, resulting in increased solvent exposure of the inhibitors. Together, these results suggest that the mutations decrease the favourability of binding by altering the dynamics of the flap regions. Notably, the insertion mutation increased PR hinge flexibility and the introduction of background mutations compensated for this by stabilising the cantilever and hinge regions. Together, these findings provide insight into how non-active site mutations affect PR conformational dynamics in critical areas of the PR thus impacting on drug binding capacity and potentially contributing to drug resistance.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"133 ","pages":"Article 108850"},"PeriodicalIF":2.7000,"publicationDate":"2024-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S1093326324001505/pdfft?md5=a95ddd1453ef958468a5ebe54b57258a&pid=1-s2.0-S1093326324001505-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Contrasting the effect of hinge region insertions and non-active site mutations on HIV protease-inhibitor interactions: Insights from altered flap dynamics\",\"authors\":\"Tshele Mokhantso, Dean Sherry, Roland Worth, Ramesh Pandian, Ikechukwu Achilonu, Yasien Sayed\",\"doi\":\"10.1016/j.jmgm.2024.108850\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>HIV-1 protease (PR) enzyme is a viable antiretroviral drug target due to its crucial role in HIV maturation. Over many decades, the HIV-1 PR enzyme has exhibited mutations brought on by drug pressure and error-prone nature of HIV-1 reverse transcriptase. Non-active site mutations have played a pivotal role in drug resistance; however, their mechanism of action has not been fully elucidated. We investigated how non-active site mutations affect the conformational stability and drug binding ability of HIV-1 PR. In light of this, we studied a novel HIV-1 subtype C protease variant containing an insertion of valine (↑V) in the hinge region. We analysed the mutations in the presence and absence of ten background mutations. Molecular dynamics simulations revealed that both with and without the background mutations, the PR exhibited increased flexibility of hinge, flaps and fulcrum regions. This allowed the PR to adopt a wider flap conformation when in complex with several inhibitors. Additionally, the simulations revealed that the protease inhibitors (PIs) could not bring the mutated variant proteases into a stable, closed conformation, resulting in increased solvent exposure of the inhibitors. Together, these results suggest that the mutations decrease the favourability of binding by altering the dynamics of the flap regions. Notably, the insertion mutation increased PR hinge flexibility and the introduction of background mutations compensated for this by stabilising the cantilever and hinge regions. Together, these findings provide insight into how non-active site mutations affect PR conformational dynamics in critical areas of the PR thus impacting on drug binding capacity and potentially contributing to drug resistance.</p></div>\",\"PeriodicalId\":16361,\"journal\":{\"name\":\"Journal of molecular graphics & modelling\",\"volume\":\"133 \",\"pages\":\"Article 108850\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-08-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S1093326324001505/pdfft?md5=a95ddd1453ef958468a5ebe54b57258a&pid=1-s2.0-S1093326324001505-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular graphics & modelling\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1093326324001505\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001505","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
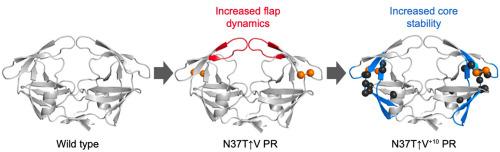
Contrasting the effect of hinge region insertions and non-active site mutations on HIV protease-inhibitor interactions: Insights from altered flap dynamics
HIV-1 protease (PR) enzyme is a viable antiretroviral drug target due to its crucial role in HIV maturation. Over many decades, the HIV-1 PR enzyme has exhibited mutations brought on by drug pressure and error-prone nature of HIV-1 reverse transcriptase. Non-active site mutations have played a pivotal role in drug resistance; however, their mechanism of action has not been fully elucidated. We investigated how non-active site mutations affect the conformational stability and drug binding ability of HIV-1 PR. In light of this, we studied a novel HIV-1 subtype C protease variant containing an insertion of valine (↑V) in the hinge region. We analysed the mutations in the presence and absence of ten background mutations. Molecular dynamics simulations revealed that both with and without the background mutations, the PR exhibited increased flexibility of hinge, flaps and fulcrum regions. This allowed the PR to adopt a wider flap conformation when in complex with several inhibitors. Additionally, the simulations revealed that the protease inhibitors (PIs) could not bring the mutated variant proteases into a stable, closed conformation, resulting in increased solvent exposure of the inhibitors. Together, these results suggest that the mutations decrease the favourability of binding by altering the dynamics of the flap regions. Notably, the insertion mutation increased PR hinge flexibility and the introduction of background mutations compensated for this by stabilising the cantilever and hinge regions. Together, these findings provide insight into how non-active site mutations affect PR conformational dynamics in critical areas of the PR thus impacting on drug binding capacity and potentially contributing to drug resistance.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


