Rhushikesh A. Phadke, Alison Brack, Luke A. Fournier, Ezra Kruzich, Mingqi Sha, Ines Picard, Connor Johnson, Dimitri Stroumbakis, Maria Salgado, Charlotte Yeung, Berta Escude Velasco, Yen Yu Liu, Alberto Cruz-Martín
{"title":"精神分裂症风险基因 C4 通过损害 AMPAR 的贩运诱导病理性突触缺失","authors":"Rhushikesh A. Phadke, Alison Brack, Luke A. Fournier, Ezra Kruzich, Mingqi Sha, Ines Picard, Connor Johnson, Dimitri Stroumbakis, Maria Salgado, Charlotte Yeung, Berta Escude Velasco, Yen Yu Liu, Alberto Cruz-Martín","doi":"10.1038/s41380-024-02701-7","DOIUrl":null,"url":null,"abstract":"<p>Neuroimmune interactions play a significant role in regulating synaptic plasticity in both the healthy and diseased brain. The complement pathway, an extracellular proteolytic cascade, exemplifies these interactions. Its activation triggers microglia-dependent synaptic elimination via the complement receptor 3 (CR3). Current models of pathological complement activity in the brain propose that accelerated synaptic loss resulting from overexpression of C4 (C4-OE), a gene associated with schizophrenia, follows this pathway. Here, we report that C4-mediated cortical hypoconnectivity is CR3-independent. Instead, C4-OE triggers impaired GluR1 trafficking through an intracellular mechanism involving the endosomal protein SNX27, resulting in pathological synaptic loss. Moreover, C4 circuit alterations in the prefrontal cortex, a brain region associated with neuropsychiatric disorders, were rescued by increasing neuronal levels of SNX27, which we identify as an interacting partner of this neuroimmune protein. Our results link excessive complement activity to an intracellular endo-lysosomal trafficking pathway altering synaptic plasticity.</p>","PeriodicalId":19008,"journal":{"name":"Molecular Psychiatry","volume":null,"pages":null},"PeriodicalIF":9.6000,"publicationDate":"2024-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The schizophrenia risk gene C4 induces pathological synaptic loss by impairing AMPAR trafficking\",\"authors\":\"Rhushikesh A. Phadke, Alison Brack, Luke A. Fournier, Ezra Kruzich, Mingqi Sha, Ines Picard, Connor Johnson, Dimitri Stroumbakis, Maria Salgado, Charlotte Yeung, Berta Escude Velasco, Yen Yu Liu, Alberto Cruz-Martín\",\"doi\":\"10.1038/s41380-024-02701-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Neuroimmune interactions play a significant role in regulating synaptic plasticity in both the healthy and diseased brain. The complement pathway, an extracellular proteolytic cascade, exemplifies these interactions. Its activation triggers microglia-dependent synaptic elimination via the complement receptor 3 (CR3). Current models of pathological complement activity in the brain propose that accelerated synaptic loss resulting from overexpression of C4 (C4-OE), a gene associated with schizophrenia, follows this pathway. Here, we report that C4-mediated cortical hypoconnectivity is CR3-independent. Instead, C4-OE triggers impaired GluR1 trafficking through an intracellular mechanism involving the endosomal protein SNX27, resulting in pathological synaptic loss. Moreover, C4 circuit alterations in the prefrontal cortex, a brain region associated with neuropsychiatric disorders, were rescued by increasing neuronal levels of SNX27, which we identify as an interacting partner of this neuroimmune protein. Our results link excessive complement activity to an intracellular endo-lysosomal trafficking pathway altering synaptic plasticity.</p>\",\"PeriodicalId\":19008,\"journal\":{\"name\":\"Molecular Psychiatry\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":9.6000,\"publicationDate\":\"2024-09-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Psychiatry\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41380-024-02701-7\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Psychiatry","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41380-024-02701-7","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
The schizophrenia risk gene C4 induces pathological synaptic loss by impairing AMPAR trafficking
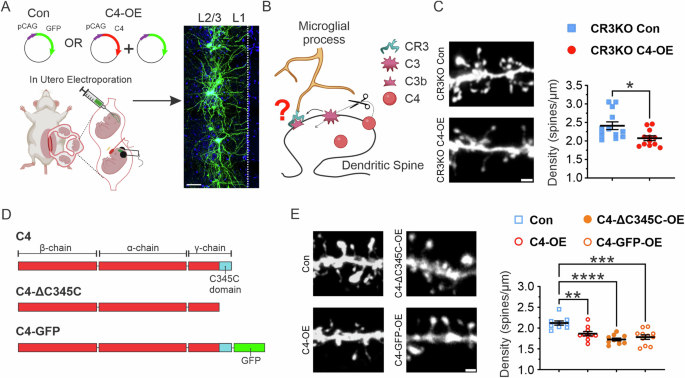
Neuroimmune interactions play a significant role in regulating synaptic plasticity in both the healthy and diseased brain. The complement pathway, an extracellular proteolytic cascade, exemplifies these interactions. Its activation triggers microglia-dependent synaptic elimination via the complement receptor 3 (CR3). Current models of pathological complement activity in the brain propose that accelerated synaptic loss resulting from overexpression of C4 (C4-OE), a gene associated with schizophrenia, follows this pathway. Here, we report that C4-mediated cortical hypoconnectivity is CR3-independent. Instead, C4-OE triggers impaired GluR1 trafficking through an intracellular mechanism involving the endosomal protein SNX27, resulting in pathological synaptic loss. Moreover, C4 circuit alterations in the prefrontal cortex, a brain region associated with neuropsychiatric disorders, were rescued by increasing neuronal levels of SNX27, which we identify as an interacting partner of this neuroimmune protein. Our results link excessive complement activity to an intracellular endo-lysosomal trafficking pathway altering synaptic plasticity.
期刊介绍:
Molecular Psychiatry focuses on publishing research that aims to uncover the biological mechanisms behind psychiatric disorders and their treatment. The journal emphasizes studies that bridge pre-clinical and clinical research, covering cellular, molecular, integrative, clinical, imaging, and psychopharmacology levels.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


