Michael A. Bellucci, Lina Yuan, Grahame R. Woollam, Bing Wang, Liwen Fang, Yunfei Zhou, Chandler Greenwell, Sivakumar Sekharan, Xiaolan Ling* and GuangXu Sun*,
{"title":"克霉唑和酮洛芬在聚合物基质上的模板成核作用","authors":"Michael A. Bellucci, Lina Yuan, Grahame R. Woollam, Bing Wang, Liwen Fang, Yunfei Zhou, Chandler Greenwell, Sivakumar Sekharan, Xiaolan Ling* and GuangXu Sun*, ","doi":"10.1021/acs.molpharmaceut.4c0049110.1021/acs.molpharmaceut.4c00491","DOIUrl":null,"url":null,"abstract":"<p >The use of different template surfaces in crystallization experiments can directly influence the nucleation kinetics, crystal growth, and morphology of active pharmaceutical ingredients (APIs). Consequently, templated nucleation is an attractive approach to enhance crystal nucleation kinetics and preferentially nucleate desired crystal polymorphs for solid-form drug molecules, particularly large and flexible molecules that are difficult to crystallize. Herein, we investigate the effect of polymer templates on the crystal nucleation of clotrimazole and ketoprofen with both experiments and computational methods. Crystallization was carried out in toluene solvent for both APIs with a template library consisting of 12 different polymers. In complement to the experimental studies, we developed a computational workflow based on molecular dynamics (MD) and derived descriptors from the simulations to score and rank API–polymer interactions. The descriptors were used to measure the energy of interaction (EOI), hydrogen bonding, and rugosity (surface roughness) similarity between the APIs and polymer templates. We used a variety of machine learning models (14 in total) along with these descriptors to predict the crystallization outcome of the polymer templates. We found that simply rank-ordering the polymers by their API–polymer interaction energy descriptors yielded 92% accuracy in predicting the experimental outcome for clotrimazole and ketoprofen. The most accurate machine learning model for both APIs was found to be a random forest model. Using these models, we were able to predict the crystallization outcomes for all polymers. Additionally, we have performed a feature importance analysis using the trained models and found that the most predictive features are the energy descriptors. These results demonstrate that API–polymer interaction energies are correlated with heterogeneous crystallization outcomes.</p>","PeriodicalId":52,"journal":{"name":"Molecular Pharmaceutics","volume":"21 9","pages":"4576–4588 4576–4588"},"PeriodicalIF":4.5000,"publicationDate":"2024-08-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Templated Nucleation of Clotrimazole and Ketoprofen on Polymer Substrates\",\"authors\":\"Michael A. Bellucci, Lina Yuan, Grahame R. Woollam, Bing Wang, Liwen Fang, Yunfei Zhou, Chandler Greenwell, Sivakumar Sekharan, Xiaolan Ling* and GuangXu Sun*, \",\"doi\":\"10.1021/acs.molpharmaceut.4c0049110.1021/acs.molpharmaceut.4c00491\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The use of different template surfaces in crystallization experiments can directly influence the nucleation kinetics, crystal growth, and morphology of active pharmaceutical ingredients (APIs). Consequently, templated nucleation is an attractive approach to enhance crystal nucleation kinetics and preferentially nucleate desired crystal polymorphs for solid-form drug molecules, particularly large and flexible molecules that are difficult to crystallize. Herein, we investigate the effect of polymer templates on the crystal nucleation of clotrimazole and ketoprofen with both experiments and computational methods. Crystallization was carried out in toluene solvent for both APIs with a template library consisting of 12 different polymers. In complement to the experimental studies, we developed a computational workflow based on molecular dynamics (MD) and derived descriptors from the simulations to score and rank API–polymer interactions. The descriptors were used to measure the energy of interaction (EOI), hydrogen bonding, and rugosity (surface roughness) similarity between the APIs and polymer templates. We used a variety of machine learning models (14 in total) along with these descriptors to predict the crystallization outcome of the polymer templates. We found that simply rank-ordering the polymers by their API–polymer interaction energy descriptors yielded 92% accuracy in predicting the experimental outcome for clotrimazole and ketoprofen. The most accurate machine learning model for both APIs was found to be a random forest model. Using these models, we were able to predict the crystallization outcomes for all polymers. Additionally, we have performed a feature importance analysis using the trained models and found that the most predictive features are the energy descriptors. These results demonstrate that API–polymer interaction energies are correlated with heterogeneous crystallization outcomes.</p>\",\"PeriodicalId\":52,\"journal\":{\"name\":\"Molecular Pharmaceutics\",\"volume\":\"21 9\",\"pages\":\"4576–4588 4576–4588\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2024-08-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Pharmaceutics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.4c00491\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Pharmaceutics","FirstCategoryId":"3","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.4c00491","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
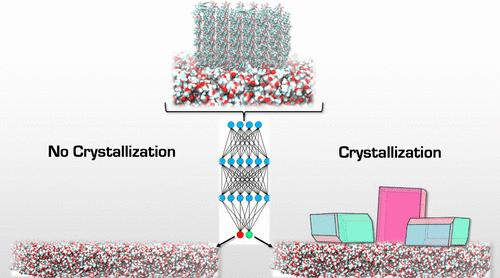
Templated Nucleation of Clotrimazole and Ketoprofen on Polymer Substrates
The use of different template surfaces in crystallization experiments can directly influence the nucleation kinetics, crystal growth, and morphology of active pharmaceutical ingredients (APIs). Consequently, templated nucleation is an attractive approach to enhance crystal nucleation kinetics and preferentially nucleate desired crystal polymorphs for solid-form drug molecules, particularly large and flexible molecules that are difficult to crystallize. Herein, we investigate the effect of polymer templates on the crystal nucleation of clotrimazole and ketoprofen with both experiments and computational methods. Crystallization was carried out in toluene solvent for both APIs with a template library consisting of 12 different polymers. In complement to the experimental studies, we developed a computational workflow based on molecular dynamics (MD) and derived descriptors from the simulations to score and rank API–polymer interactions. The descriptors were used to measure the energy of interaction (EOI), hydrogen bonding, and rugosity (surface roughness) similarity between the APIs and polymer templates. We used a variety of machine learning models (14 in total) along with these descriptors to predict the crystallization outcome of the polymer templates. We found that simply rank-ordering the polymers by their API–polymer interaction energy descriptors yielded 92% accuracy in predicting the experimental outcome for clotrimazole and ketoprofen. The most accurate machine learning model for both APIs was found to be a random forest model. Using these models, we were able to predict the crystallization outcomes for all polymers. Additionally, we have performed a feature importance analysis using the trained models and found that the most predictive features are the energy descriptors. These results demonstrate that API–polymer interaction energies are correlated with heterogeneous crystallization outcomes.
期刊介绍:
Molecular Pharmaceutics publishes the results of original research that contributes significantly to the molecular mechanistic understanding of drug delivery and drug delivery systems. The journal encourages contributions describing research at the interface of drug discovery and drug development.
Scientific areas within the scope of the journal include physical and pharmaceutical chemistry, biochemistry and biophysics, molecular and cellular biology, and polymer and materials science as they relate to drug and drug delivery system efficacy. Mechanistic Drug Delivery and Drug Targeting research on modulating activity and efficacy of a drug or drug product is within the scope of Molecular Pharmaceutics. Theoretical and experimental peer-reviewed research articles, communications, reviews, and perspectives are welcomed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


