Elijah T Roberts, Alexander R Davis, Jeremy T Risher, Adam W Barb, I Jonathan Amster
{"title":"自动分配双标记蛋白质中的 15N 和 13C 富集水平","authors":"Elijah T Roberts, Alexander R Davis, Jeremy T Risher, Adam W Barb, I Jonathan Amster","doi":"10.1021/jasms.4c00218","DOIUrl":null,"url":null,"abstract":"<p><p>Uniform enrichment of <sup>15</sup>N and <sup>13</sup>C in proteins is commonly employed for 2D heteronuclear NMR measurements of the three-dimensional protein structure. Achieving a high degree of enrichment of both elements is important for obtaining high quality data. Uniform labeling of proteins and glycoproteins expressed in higher organisms (yeast or mammalian cell lines) is more challenging than expression in <i>Escherichia coli</i>, a prokaryote that grows on simple, chemically defined media but does not provide appropriate eukaryotic modifications. It is difficult to achieve complete incorporation of both heavy isotopes, and quality control measures are important for quantitating the level of their enrichment. Mass spectrometry measurements of the isotopic distribution of the intact protein or its proteolytic fragments provide the means to assess the enrichment level. A mass accuracy of 1 ppm or better is shown to be required to distinguish the correct combination of <sup>13</sup>C and <sup>15</sup>N enrichment due to subtle shifts in peak centroids with differences in the underlying, but unresolved, isotopic fine structure. A simple computer program was developed to optimize the fitting of experimental isotope patterns to statistically derived distributions. This method can determine the isotopic abundance from isotope patterns and isotopologue masses in conventional MS data for peptides, intact proteins, and glycans. For this purpose, MATLAB's isotope simulator, isotopicdist, has been modified to permit the variation of <sup>15</sup>N and <sup>13</sup>C enrichment levels and to perform a two-dimensional grid search of enrichment levels of both isotopes. We also incorporated an alternate isotope simulator, js-emass, into MATLAB for use in the same fitting program. Herein we benchmark this technique on natural abundance ubiquitin and uniformly [<sup>15</sup>N,<sup>13</sup>C]-labeled ubiquitin at both the intact and peptide level, outline considerations for data quality and mass accuracy, and report several improvements we have made to the previously reported analysis of the [<sup>15</sup>N,<sup>13</sup>C]-enriched human IgG Fc domain, a glycoprotein that has been expressed in <i>Saccharomyces cerevisiae</i>.</p>","PeriodicalId":672,"journal":{"name":"Journal of the American Society for Mass Spectrometry","volume":null,"pages":null},"PeriodicalIF":3.1000,"publicationDate":"2024-10-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11450805/pdf/","citationCount":"0","resultStr":"{\"title\":\"Automated Assignment of <sup>15</sup>N And <sup>13</sup>C Enrichment Levels in Doubly-Labeled Proteins.\",\"authors\":\"Elijah T Roberts, Alexander R Davis, Jeremy T Risher, Adam W Barb, I Jonathan Amster\",\"doi\":\"10.1021/jasms.4c00218\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Uniform enrichment of <sup>15</sup>N and <sup>13</sup>C in proteins is commonly employed for 2D heteronuclear NMR measurements of the three-dimensional protein structure. Achieving a high degree of enrichment of both elements is important for obtaining high quality data. Uniform labeling of proteins and glycoproteins expressed in higher organisms (yeast or mammalian cell lines) is more challenging than expression in <i>Escherichia coli</i>, a prokaryote that grows on simple, chemically defined media but does not provide appropriate eukaryotic modifications. It is difficult to achieve complete incorporation of both heavy isotopes, and quality control measures are important for quantitating the level of their enrichment. Mass spectrometry measurements of the isotopic distribution of the intact protein or its proteolytic fragments provide the means to assess the enrichment level. A mass accuracy of 1 ppm or better is shown to be required to distinguish the correct combination of <sup>13</sup>C and <sup>15</sup>N enrichment due to subtle shifts in peak centroids with differences in the underlying, but unresolved, isotopic fine structure. A simple computer program was developed to optimize the fitting of experimental isotope patterns to statistically derived distributions. This method can determine the isotopic abundance from isotope patterns and isotopologue masses in conventional MS data for peptides, intact proteins, and glycans. For this purpose, MATLAB's isotope simulator, isotopicdist, has been modified to permit the variation of <sup>15</sup>N and <sup>13</sup>C enrichment levels and to perform a two-dimensional grid search of enrichment levels of both isotopes. We also incorporated an alternate isotope simulator, js-emass, into MATLAB for use in the same fitting program. Herein we benchmark this technique on natural abundance ubiquitin and uniformly [<sup>15</sup>N,<sup>13</sup>C]-labeled ubiquitin at both the intact and peptide level, outline considerations for data quality and mass accuracy, and report several improvements we have made to the previously reported analysis of the [<sup>15</sup>N,<sup>13</sup>C]-enriched human IgG Fc domain, a glycoprotein that has been expressed in <i>Saccharomyces cerevisiae</i>.</p>\",\"PeriodicalId\":672,\"journal\":{\"name\":\"Journal of the American Society for Mass Spectrometry\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-10-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11450805/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the American Society for Mass Spectrometry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/jasms.4c00218\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/30 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Society for Mass Spectrometry","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/jasms.4c00218","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/30 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Automated Assignment of 15N And 13C Enrichment Levels in Doubly-Labeled Proteins.
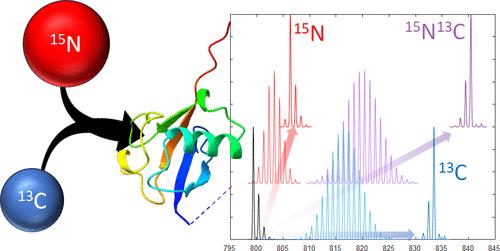
Uniform enrichment of 15N and 13C in proteins is commonly employed for 2D heteronuclear NMR measurements of the three-dimensional protein structure. Achieving a high degree of enrichment of both elements is important for obtaining high quality data. Uniform labeling of proteins and glycoproteins expressed in higher organisms (yeast or mammalian cell lines) is more challenging than expression in Escherichia coli, a prokaryote that grows on simple, chemically defined media but does not provide appropriate eukaryotic modifications. It is difficult to achieve complete incorporation of both heavy isotopes, and quality control measures are important for quantitating the level of their enrichment. Mass spectrometry measurements of the isotopic distribution of the intact protein or its proteolytic fragments provide the means to assess the enrichment level. A mass accuracy of 1 ppm or better is shown to be required to distinguish the correct combination of 13C and 15N enrichment due to subtle shifts in peak centroids with differences in the underlying, but unresolved, isotopic fine structure. A simple computer program was developed to optimize the fitting of experimental isotope patterns to statistically derived distributions. This method can determine the isotopic abundance from isotope patterns and isotopologue masses in conventional MS data for peptides, intact proteins, and glycans. For this purpose, MATLAB's isotope simulator, isotopicdist, has been modified to permit the variation of 15N and 13C enrichment levels and to perform a two-dimensional grid search of enrichment levels of both isotopes. We also incorporated an alternate isotope simulator, js-emass, into MATLAB for use in the same fitting program. Herein we benchmark this technique on natural abundance ubiquitin and uniformly [15N,13C]-labeled ubiquitin at both the intact and peptide level, outline considerations for data quality and mass accuracy, and report several improvements we have made to the previously reported analysis of the [15N,13C]-enriched human IgG Fc domain, a glycoprotein that has been expressed in Saccharomyces cerevisiae.
期刊介绍:
The Journal of the American Society for Mass Spectrometry presents research papers covering all aspects of mass spectrometry, incorporating coverage of fields of scientific inquiry in which mass spectrometry can play a role.
Comprehensive in scope, the journal publishes papers on both fundamentals and applications of mass spectrometry. Fundamental subjects include instrumentation principles, design, and demonstration, structures and chemical properties of gas-phase ions, studies of thermodynamic properties, ion spectroscopy, chemical kinetics, mechanisms of ionization, theories of ion fragmentation, cluster ions, and potential energy surfaces. In addition to full papers, the journal offers Communications, Application Notes, and Accounts and Perspectives

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


