{"title":"Kohn-Sham 密度函数对主族金属和过渡金属反应的精确度有多高","authors":"Yizhen Wang, Igor Ying Zhang, Xin Xu","doi":"10.1002/jcc.27488","DOIUrl":null,"url":null,"abstract":"<p>Achieving chemical accuracy in describing reactions involving both main-group elements and transition metals poses a substantial challenge for density functional approximations (DFAs), primarily due to the significantly different behaviors for electrons moving in the s,p-orbitals or in the d,f-orbitals. MOR41, a representative dataset of transition metal chemistry, has highlighted the PWPB95-D3(BJ) functional, a B2PLYP-type doubly hybrid (bDH) approximation equipped with an empirical dispersion correction, as the leading functional thus far (Dohm et al., J Chem Theory Comput 2018;14: 2596–2608). However, this functional is not among the top bDH methods for main-group chemistry (Goerigk et al., Phys Chem Chem Phys. 2017;19: 32184). Conversely, bDH methods such as DSD-BLYP-D3, proficient in main-group chemistry, often falter for transition metal chemistry. Herein, taking advantage of the home-made Rust-based Electronic-Structure Toolkits, we examine a suite of XYG3-type doubly hybrid (xDH) methods. We confirm that the trade-off in descriptive accuracy between main-group and transition metal systems persists within the realm of perturbation theory (PT2)-based xDH methods. Notably, however, our study ushers in a pivotal advance with the recently proposed renormalized xDH method, R-xDH7-SCC15. This method not only distinguishes itself among the elite methods for main-group chemistry, but also achieves an unprecedented accuracy for the MOR41 dataset, outperforming all other reported DFAs. The efficacy of R-xDH7-SCC15 stems from the successful integration of a renormalized PT2 correlation model (rPT2) and a machine-learning strong-correlation correction (SCC15), marking a significant step forward in the realm of computational chemistry.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2878-2884"},"PeriodicalIF":3.4000,"publicationDate":"2024-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"How accurate can Kohn-Sham density functional be for both main-group and transition metal reactions\",\"authors\":\"Yizhen Wang, Igor Ying Zhang, Xin Xu\",\"doi\":\"10.1002/jcc.27488\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Achieving chemical accuracy in describing reactions involving both main-group elements and transition metals poses a substantial challenge for density functional approximations (DFAs), primarily due to the significantly different behaviors for electrons moving in the s,p-orbitals or in the d,f-orbitals. MOR41, a representative dataset of transition metal chemistry, has highlighted the PWPB95-D3(BJ) functional, a B2PLYP-type doubly hybrid (bDH) approximation equipped with an empirical dispersion correction, as the leading functional thus far (Dohm et al., J Chem Theory Comput 2018;14: 2596–2608). However, this functional is not among the top bDH methods for main-group chemistry (Goerigk et al., Phys Chem Chem Phys. 2017;19: 32184). Conversely, bDH methods such as DSD-BLYP-D3, proficient in main-group chemistry, often falter for transition metal chemistry. Herein, taking advantage of the home-made Rust-based Electronic-Structure Toolkits, we examine a suite of XYG3-type doubly hybrid (xDH) methods. We confirm that the trade-off in descriptive accuracy between main-group and transition metal systems persists within the realm of perturbation theory (PT2)-based xDH methods. Notably, however, our study ushers in a pivotal advance with the recently proposed renormalized xDH method, R-xDH7-SCC15. This method not only distinguishes itself among the elite methods for main-group chemistry, but also achieves an unprecedented accuracy for the MOR41 dataset, outperforming all other reported DFAs. The efficacy of R-xDH7-SCC15 stems from the successful integration of a renormalized PT2 correlation model (rPT2) and a machine-learning strong-correlation correction (SCC15), marking a significant step forward in the realm of computational chemistry.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 32\",\"pages\":\"2878-2884\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-08-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27488\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27488","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
How accurate can Kohn-Sham density functional be for both main-group and transition metal reactions
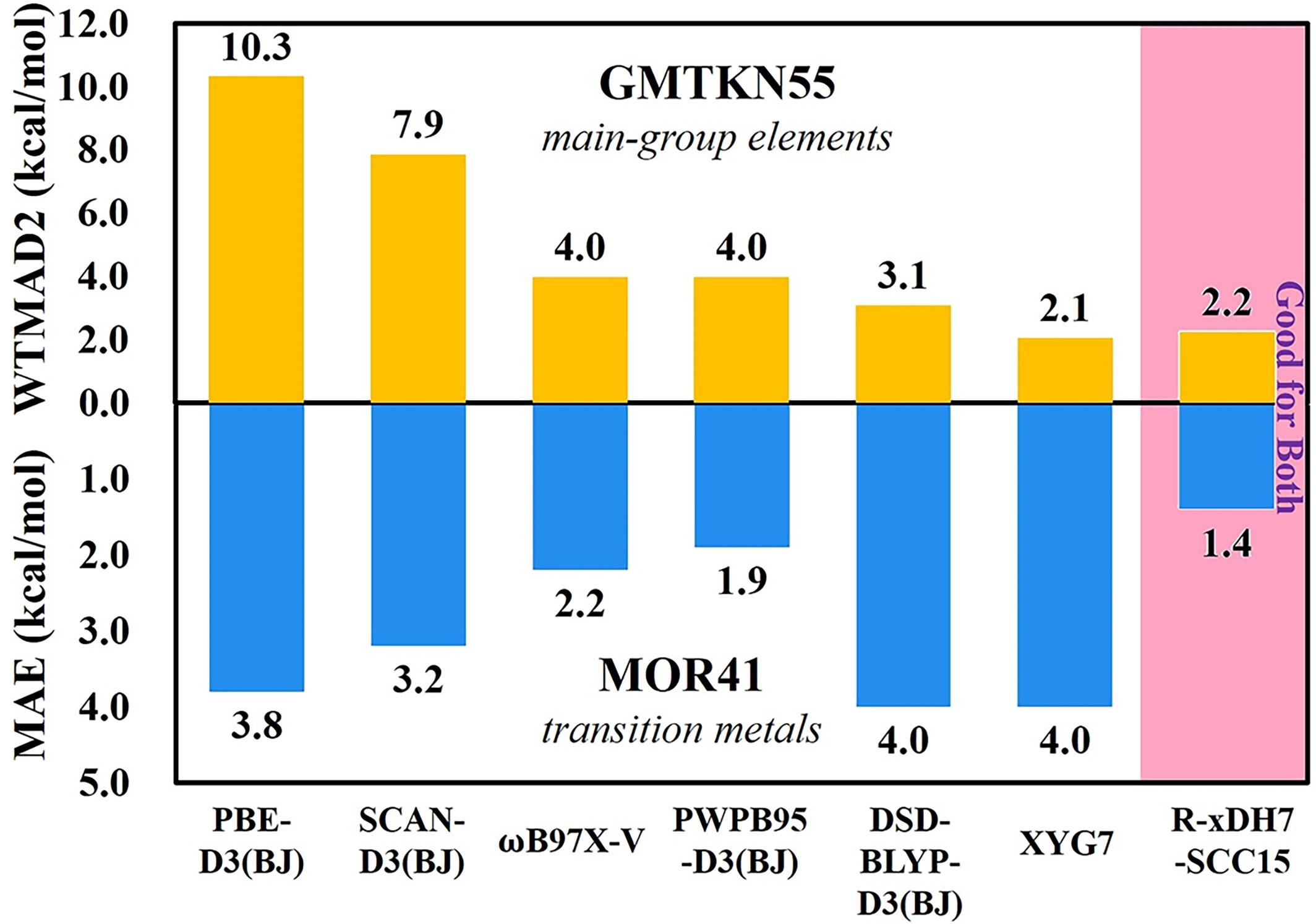
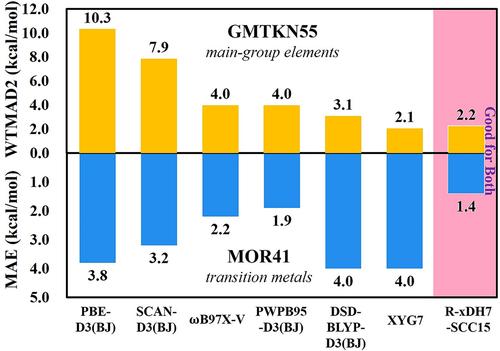
Achieving chemical accuracy in describing reactions involving both main-group elements and transition metals poses a substantial challenge for density functional approximations (DFAs), primarily due to the significantly different behaviors for electrons moving in the s,p-orbitals or in the d,f-orbitals. MOR41, a representative dataset of transition metal chemistry, has highlighted the PWPB95-D3(BJ) functional, a B2PLYP-type doubly hybrid (bDH) approximation equipped with an empirical dispersion correction, as the leading functional thus far (Dohm et al., J Chem Theory Comput 2018;14: 2596–2608). However, this functional is not among the top bDH methods for main-group chemistry (Goerigk et al., Phys Chem Chem Phys. 2017;19: 32184). Conversely, bDH methods such as DSD-BLYP-D3, proficient in main-group chemistry, often falter for transition metal chemistry. Herein, taking advantage of the home-made Rust-based Electronic-Structure Toolkits, we examine a suite of XYG3-type doubly hybrid (xDH) methods. We confirm that the trade-off in descriptive accuracy between main-group and transition metal systems persists within the realm of perturbation theory (PT2)-based xDH methods. Notably, however, our study ushers in a pivotal advance with the recently proposed renormalized xDH method, R-xDH7-SCC15. This method not only distinguishes itself among the elite methods for main-group chemistry, but also achieves an unprecedented accuracy for the MOR41 dataset, outperforming all other reported DFAs. The efficacy of R-xDH7-SCC15 stems from the successful integration of a renormalized PT2 correlation model (rPT2) and a machine-learning strong-correlation correction (SCC15), marking a significant step forward in the realm of computational chemistry.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


