{"title":"空间转录组学并不需要建立零膨胀模型。","authors":"Peiyao Zhao, Jiaqiang Zhu, Ying Ma, Xiang Zhou","doi":"10.1186/s13059-022-02684-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Spatial transcriptomics are a set of new technologies that profile gene expression on tissues with spatial localization information. With technological advances, recent spatial transcriptomics data are often in the form of sparse counts with an excessive amount of zero values.</p><p><strong>Results: </strong>We perform a comprehensive analysis on 20 spatial transcriptomics datasets collected from 11 distinct technologies to characterize the distributional properties of the expression count data and understand the statistical nature of the zero values. Across datasets, we show that a substantial fraction of genes displays overdispersion and/or zero inflation that cannot be accounted for by a Poisson model, with genes displaying overdispersion substantially overlapped with genes displaying zero inflation. In addition, we find that either the Poisson or the negative binomial model is sufficient for modeling the majority of genes across most spatial transcriptomics technologies. We further show major sources of overdispersion and zero inflation in spatial transcriptomics including gene expression heterogeneity across tissue locations and spatial distribution of cell types. In particular, when we focus on a relatively homogeneous set of tissue locations or control for cell type compositions, the number of detected overdispersed and/or zero-inflated genes is substantially reduced, and a simple Poisson model is often sufficient to fit the gene expression data there.</p><p><strong>Conclusions: </strong>Our study provides the first comprehensive evidence that excessive zeros in spatial transcriptomics are not due to zero inflation, supporting the use of count models without a zero inflation component for modeling spatial transcriptomics.</p>","PeriodicalId":48922,"journal":{"name":"Genome Biology","volume":"23 1","pages":"118"},"PeriodicalIF":12.3000,"publicationDate":"2022-05-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9116027/pdf/","citationCount":"0","resultStr":"{\"title\":\"Modeling zero inflation is not necessary for spatial transcriptomics.\",\"authors\":\"Peiyao Zhao, Jiaqiang Zhu, Ying Ma, Xiang Zhou\",\"doi\":\"10.1186/s13059-022-02684-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Spatial transcriptomics are a set of new technologies that profile gene expression on tissues with spatial localization information. With technological advances, recent spatial transcriptomics data are often in the form of sparse counts with an excessive amount of zero values.</p><p><strong>Results: </strong>We perform a comprehensive analysis on 20 spatial transcriptomics datasets collected from 11 distinct technologies to characterize the distributional properties of the expression count data and understand the statistical nature of the zero values. Across datasets, we show that a substantial fraction of genes displays overdispersion and/or zero inflation that cannot be accounted for by a Poisson model, with genes displaying overdispersion substantially overlapped with genes displaying zero inflation. In addition, we find that either the Poisson or the negative binomial model is sufficient for modeling the majority of genes across most spatial transcriptomics technologies. We further show major sources of overdispersion and zero inflation in spatial transcriptomics including gene expression heterogeneity across tissue locations and spatial distribution of cell types. In particular, when we focus on a relatively homogeneous set of tissue locations or control for cell type compositions, the number of detected overdispersed and/or zero-inflated genes is substantially reduced, and a simple Poisson model is often sufficient to fit the gene expression data there.</p><p><strong>Conclusions: </strong>Our study provides the first comprehensive evidence that excessive zeros in spatial transcriptomics are not due to zero inflation, supporting the use of count models without a zero inflation component for modeling spatial transcriptomics.</p>\",\"PeriodicalId\":48922,\"journal\":{\"name\":\"Genome Biology\",\"volume\":\"23 1\",\"pages\":\"118\"},\"PeriodicalIF\":12.3000,\"publicationDate\":\"2022-05-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9116027/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genome Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13059-022-02684-0\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genome Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13059-022-02684-0","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
Modeling zero inflation is not necessary for spatial transcriptomics.
Background: Spatial transcriptomics are a set of new technologies that profile gene expression on tissues with spatial localization information. With technological advances, recent spatial transcriptomics data are often in the form of sparse counts with an excessive amount of zero values.
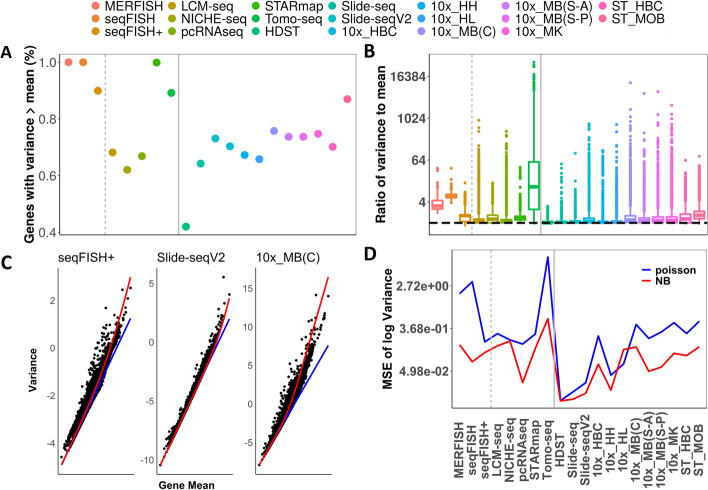
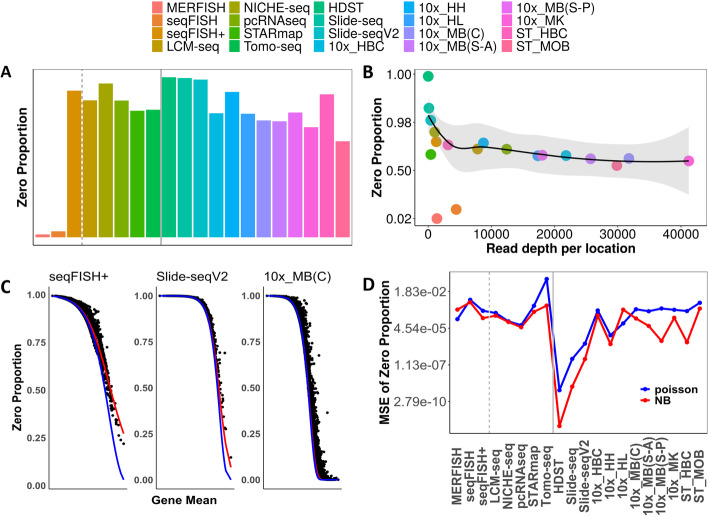
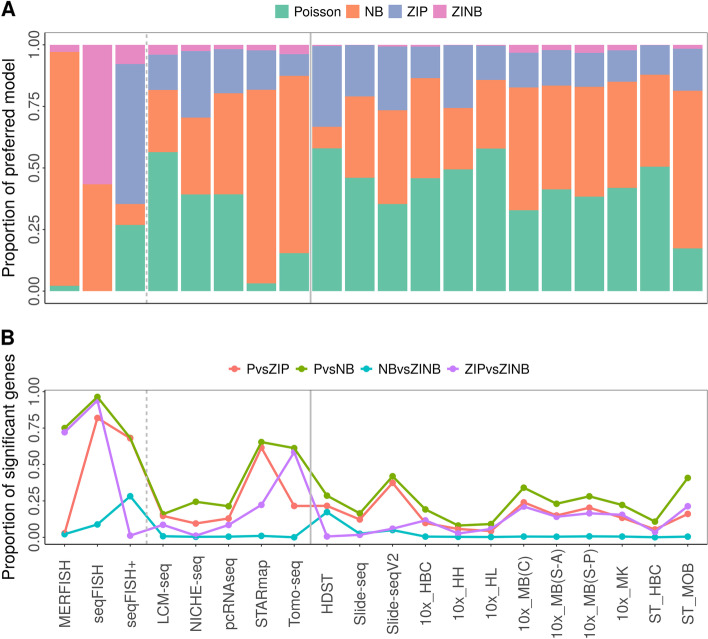
Results: We perform a comprehensive analysis on 20 spatial transcriptomics datasets collected from 11 distinct technologies to characterize the distributional properties of the expression count data and understand the statistical nature of the zero values. Across datasets, we show that a substantial fraction of genes displays overdispersion and/or zero inflation that cannot be accounted for by a Poisson model, with genes displaying overdispersion substantially overlapped with genes displaying zero inflation. In addition, we find that either the Poisson or the negative binomial model is sufficient for modeling the majority of genes across most spatial transcriptomics technologies. We further show major sources of overdispersion and zero inflation in spatial transcriptomics including gene expression heterogeneity across tissue locations and spatial distribution of cell types. In particular, when we focus on a relatively homogeneous set of tissue locations or control for cell type compositions, the number of detected overdispersed and/or zero-inflated genes is substantially reduced, and a simple Poisson model is often sufficient to fit the gene expression data there.
Conclusions: Our study provides the first comprehensive evidence that excessive zeros in spatial transcriptomics are not due to zero inflation, supporting the use of count models without a zero inflation component for modeling spatial transcriptomics.
期刊介绍:
Genome Biology is a leading research journal that focuses on the study of biology and biomedicine from a genomic and post-genomic standpoint. The journal consistently publishes outstanding research across various areas within these fields.
With an impressive impact factor of 12.3 (2022), Genome Biology has earned its place as the 3rd highest-ranked research journal in the Genetics and Heredity category, according to Thomson Reuters. Additionally, it is ranked 2nd among research journals in the Biotechnology and Applied Microbiology category. It is important to note that Genome Biology is the top-ranking open access journal in this category.
In summary, Genome Biology sets a high standard for scientific publications in the field, showcasing cutting-edge research and earning recognition among its peers.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


