Md Arifur Rahman, Ardeshir Amirkhani, Maria Mempin, Seong Beom Ahn, Anand K Deva, Mark S Baker, Karen Vickery, Honghua Hu
{"title":"低丰度血浆蛋白质组揭示了与乳房假体囊性挛缩相关的不同丰度蛋白质:一项试点研究。","authors":"Md Arifur Rahman, Ardeshir Amirkhani, Maria Mempin, Seong Beom Ahn, Anand K Deva, Mark S Baker, Karen Vickery, Honghua Hu","doi":"10.3390/proteomes12030022","DOIUrl":null,"url":null,"abstract":"<p><p>Capsular contracture (CC) is one of the most common postoperative complications associated with breast implant-associated infections. The mechanisms that lead to CC remain poorly understood. Plasma is an ideal biospecimen for early proteomics biomarker discovery. However, as high-abundance proteins mask signals from low-abundance proteins, identifying novel or specific proteins as biomarkers for a particular disease has been hampered. Here, we employed depletion of high-abundance plasma proteins followed by Tandem Mass Tag (TMT)-based quantitative proteomics to compare 10 healthy control patients against 10 breast implant CC patients. A total of 450 proteins were identified from these samples. Among them, 16 proteins were significantly differentially expressed in which 5 proteins were upregulated and 11 downregulated in breast implant CC patients compared to healthy controls. Gene Ontology enrichment analysis revealed that proteins related to cell, cellular processes and catalytic activity were highest in the cellular component, biological process, and molecular function categories, respectively. Further, pathway analysis revealed that inflammatory responses, focal adhesion, platelet activation, and complement and coagulation cascades were enriched pathways. The differentially abundant proteins from TMT-based quantitative proteomics have the potential to provide important information for future mechanistic studies and in the development of breast implant CC biomarkers.</p>","PeriodicalId":20877,"journal":{"name":"Proteomes","volume":"12 3","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2024-08-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11348101/pdf/","citationCount":"0","resultStr":"{\"title\":\"The Low-Abundance Plasma Proteome Reveals Differentially Abundant Proteins Associated with Breast Implant Capsular Contracture: A Pilot Study.\",\"authors\":\"Md Arifur Rahman, Ardeshir Amirkhani, Maria Mempin, Seong Beom Ahn, Anand K Deva, Mark S Baker, Karen Vickery, Honghua Hu\",\"doi\":\"10.3390/proteomes12030022\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Capsular contracture (CC) is one of the most common postoperative complications associated with breast implant-associated infections. The mechanisms that lead to CC remain poorly understood. Plasma is an ideal biospecimen for early proteomics biomarker discovery. However, as high-abundance proteins mask signals from low-abundance proteins, identifying novel or specific proteins as biomarkers for a particular disease has been hampered. Here, we employed depletion of high-abundance plasma proteins followed by Tandem Mass Tag (TMT)-based quantitative proteomics to compare 10 healthy control patients against 10 breast implant CC patients. A total of 450 proteins were identified from these samples. Among them, 16 proteins were significantly differentially expressed in which 5 proteins were upregulated and 11 downregulated in breast implant CC patients compared to healthy controls. Gene Ontology enrichment analysis revealed that proteins related to cell, cellular processes and catalytic activity were highest in the cellular component, biological process, and molecular function categories, respectively. Further, pathway analysis revealed that inflammatory responses, focal adhesion, platelet activation, and complement and coagulation cascades were enriched pathways. The differentially abundant proteins from TMT-based quantitative proteomics have the potential to provide important information for future mechanistic studies and in the development of breast implant CC biomarkers.</p>\",\"PeriodicalId\":20877,\"journal\":{\"name\":\"Proteomes\",\"volume\":\"12 3\",\"pages\":\"\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2024-08-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11348101/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Proteomes\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/proteomes12030022\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomes","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/proteomes12030022","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
囊性挛缩(CC)是乳房植入物相关感染最常见的术后并发症之一。人们对导致囊性挛缩的机制仍然知之甚少。血浆是发现早期蛋白质组学生物标志物的理想生物样本。然而,由于高丰度蛋白质会掩盖低丰度蛋白质的信号,因此将新型或特异性蛋白质鉴定为特定疾病的生物标记物一直受到阻碍。在此,我们采用去高丰度血浆蛋白,然后基于串联质量标签(TMT)的定量蛋白质组学方法,比较了 10 名健康对照组患者和 10 名乳房植入物 CC 患者。从这些样本中共鉴定出 450 种蛋白质。其中,与健康对照组相比,乳房植入物 CC 患者的 16 个蛋白质有明显的差异表达,其中 5 个蛋白质上调,11 个蛋白质下调。基因本体富集分析显示,与细胞、细胞过程和催化活性有关的蛋白质分别在细胞成分、生物过程和分子功能类别中含量最高。此外,通路分析显示,炎症反应、病灶粘附、血小板活化以及补体和凝血级联是富集的通路。基于 TMT 的定量蛋白质组学研究得出的差异丰度蛋白有可能为未来的机理研究和乳房植入物 CC 生物标记物的开发提供重要信息。
The Low-Abundance Plasma Proteome Reveals Differentially Abundant Proteins Associated with Breast Implant Capsular Contracture: A Pilot Study.
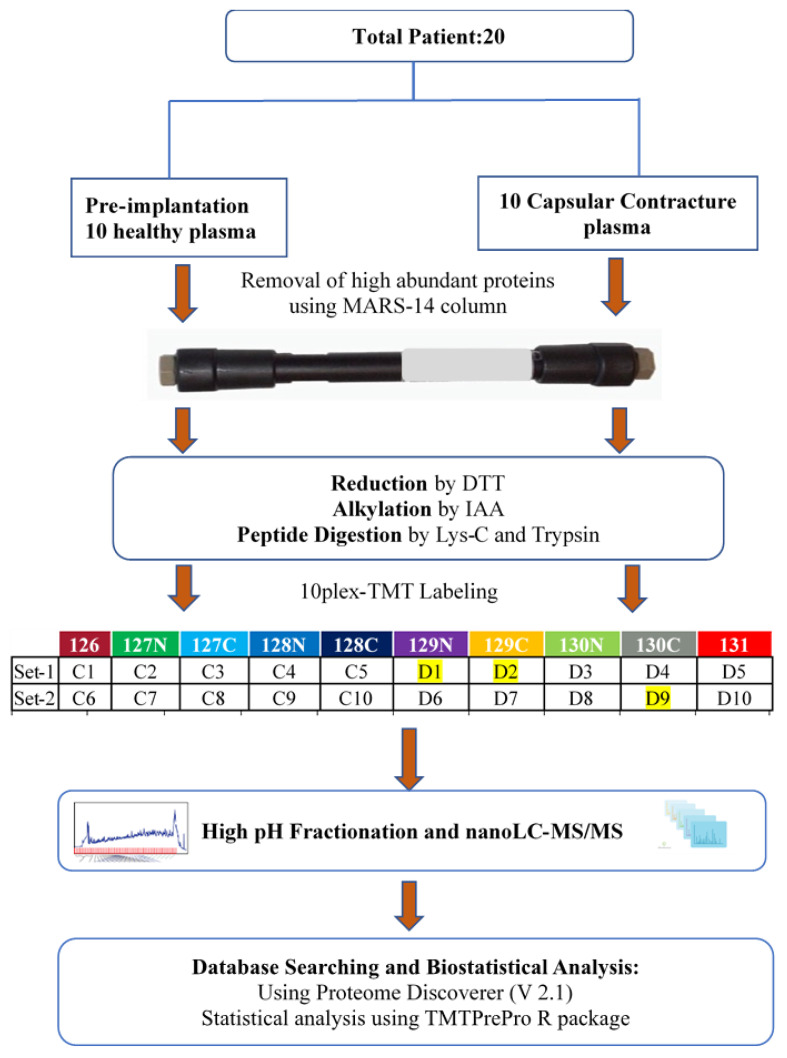
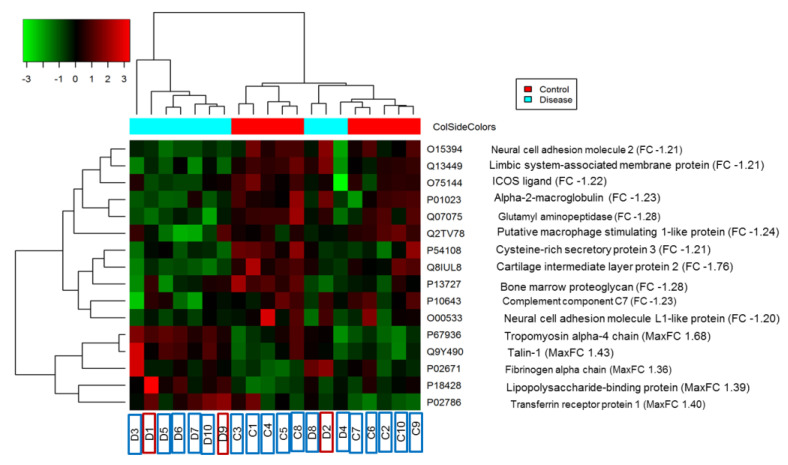
Capsular contracture (CC) is one of the most common postoperative complications associated with breast implant-associated infections. The mechanisms that lead to CC remain poorly understood. Plasma is an ideal biospecimen for early proteomics biomarker discovery. However, as high-abundance proteins mask signals from low-abundance proteins, identifying novel or specific proteins as biomarkers for a particular disease has been hampered. Here, we employed depletion of high-abundance plasma proteins followed by Tandem Mass Tag (TMT)-based quantitative proteomics to compare 10 healthy control patients against 10 breast implant CC patients. A total of 450 proteins were identified from these samples. Among them, 16 proteins were significantly differentially expressed in which 5 proteins were upregulated and 11 downregulated in breast implant CC patients compared to healthy controls. Gene Ontology enrichment analysis revealed that proteins related to cell, cellular processes and catalytic activity were highest in the cellular component, biological process, and molecular function categories, respectively. Further, pathway analysis revealed that inflammatory responses, focal adhesion, platelet activation, and complement and coagulation cascades were enriched pathways. The differentially abundant proteins from TMT-based quantitative proteomics have the potential to provide important information for future mechanistic studies and in the development of breast implant CC biomarkers.
ProteomesBiochemistry, Genetics and Molecular Biology-Clinical Biochemistry
CiteScore
6.50
自引率
3.00%
发文量
37
审稿时长
11 weeks
期刊介绍:
Proteomes (ISSN 2227-7382) is an open access, peer reviewed journal on all aspects of proteome science. Proteomes covers the multi-disciplinary topics of structural and functional biology, protein chemistry, cell biology, methodology used for protein analysis, including mass spectrometry, protein arrays, bioinformatics, HTS assays, etc. Our aim is to encourage scientists to publish their experimental and theoretical results in as much detail as possible. Therefore, there is no restriction on the length of papers. Scope: -whole proteome analysis of any organism -disease/pharmaceutical studies -comparative proteomics -protein-ligand/protein interactions -structure/functional proteomics -gene expression -methodology -bioinformatics -applications of proteomics

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


