Clarisse Gotti, Florence Roux-Dalvai, Ève Bérubé, Antoine Lacombe-Rastoll, Mickaël Leclercq, Cristina C Jacob, Maurice Boissinot, Claudia Martins, Neloni R Wijeratne, Michel G Bergeron, Arnaud Droit
{"title":"LC-SRM 与机器学习相结合,能够快速鉴定和定量尿路感染中的细菌病原体。","authors":"Clarisse Gotti, Florence Roux-Dalvai, Ève Bérubé, Antoine Lacombe-Rastoll, Mickaël Leclercq, Cristina C Jacob, Maurice Boissinot, Claudia Martins, Neloni R Wijeratne, Michel G Bergeron, Arnaud Droit","doi":"10.1016/j.mcpro.2024.100832","DOIUrl":null,"url":null,"abstract":"<p><p>Urinary tract infections (UTIs) are a worldwide health problem. Fast and accurate detection of bacterial infection is essential to provide appropriate antibiotherapy to patients and to avoid the emergence of drug-resistant pathogens. While the gold standard requires 24 h to 48 h of bacteria culture prior to MALDI-TOF species identification, we propose a culture-free workflow, enabling bacterial identification and quantification in less than 4 h using 1 ml of urine. After rapid and automatable sample preparation, a signature of 82 bacterial peptides, defined by machine learning, was monitored in LC-MS, to distinguish the 15 species causing 84% of the UTIs. The combination of the sensitivity of the SRM mode on a triple quadrupole TSQ Altis instrument and the robustness of capillary flow enabled us to analyze up to 75 samples per day, with 99.2% accuracy on bacterial inoculations of healthy urines. We have also shown our method can be used to quantify the spread of the infection, from 8 × 10<sup>4</sup> to 3 × 10<sup>7</sup> CFU/ml. Finally, the workflow was validated on 45 inoculated urines and on 84 UTI-positive urine from patients, with respectively 93.3% and 87.1% of agreement with the culture-MALDI procedure at a level above 1 × 10<sup>5</sup> CFU/ml corresponding to an infection requiring antibiotherapy.</p>","PeriodicalId":18712,"journal":{"name":"Molecular & Cellular Proteomics","volume":" ","pages":"100832"},"PeriodicalIF":5.5000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11532907/pdf/","citationCount":"0","resultStr":"{\"title\":\"LC-SRM Combined With Machine Learning Enables Fast Identification and Quantification of Bacterial Pathogens in Urinary Tract Infections.\",\"authors\":\"Clarisse Gotti, Florence Roux-Dalvai, Ève Bérubé, Antoine Lacombe-Rastoll, Mickaël Leclercq, Cristina C Jacob, Maurice Boissinot, Claudia Martins, Neloni R Wijeratne, Michel G Bergeron, Arnaud Droit\",\"doi\":\"10.1016/j.mcpro.2024.100832\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Urinary tract infections (UTIs) are a worldwide health problem. Fast and accurate detection of bacterial infection is essential to provide appropriate antibiotherapy to patients and to avoid the emergence of drug-resistant pathogens. While the gold standard requires 24 h to 48 h of bacteria culture prior to MALDI-TOF species identification, we propose a culture-free workflow, enabling bacterial identification and quantification in less than 4 h using 1 ml of urine. After rapid and automatable sample preparation, a signature of 82 bacterial peptides, defined by machine learning, was monitored in LC-MS, to distinguish the 15 species causing 84% of the UTIs. The combination of the sensitivity of the SRM mode on a triple quadrupole TSQ Altis instrument and the robustness of capillary flow enabled us to analyze up to 75 samples per day, with 99.2% accuracy on bacterial inoculations of healthy urines. We have also shown our method can be used to quantify the spread of the infection, from 8 × 10<sup>4</sup> to 3 × 10<sup>7</sup> CFU/ml. Finally, the workflow was validated on 45 inoculated urines and on 84 UTI-positive urine from patients, with respectively 93.3% and 87.1% of agreement with the culture-MALDI procedure at a level above 1 × 10<sup>5</sup> CFU/ml corresponding to an infection requiring antibiotherapy.</p>\",\"PeriodicalId\":18712,\"journal\":{\"name\":\"Molecular & Cellular Proteomics\",\"volume\":\" \",\"pages\":\"100832\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11532907/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular & Cellular Proteomics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1016/j.mcpro.2024.100832\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/22 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular & Cellular Proteomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.mcpro.2024.100832","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/22 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
LC-SRM Combined With Machine Learning Enables Fast Identification and Quantification of Bacterial Pathogens in Urinary Tract Infections.
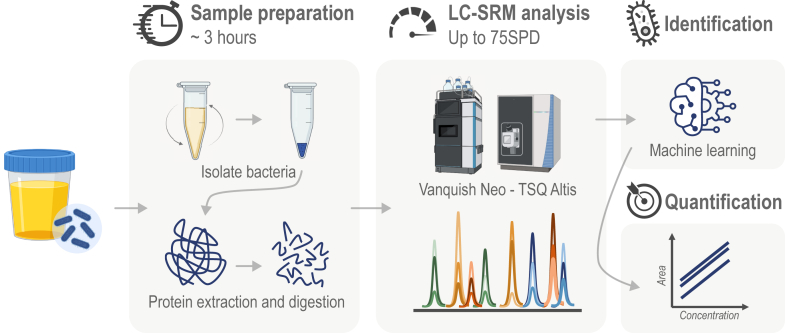
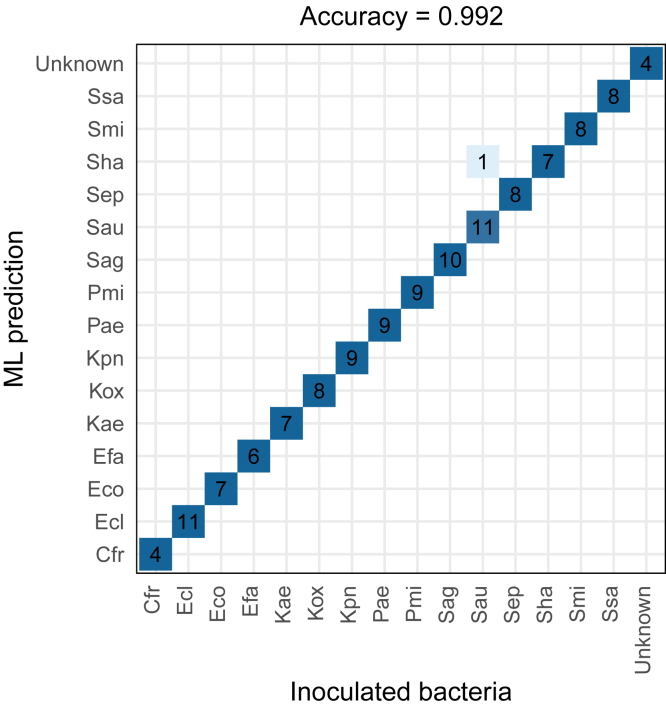
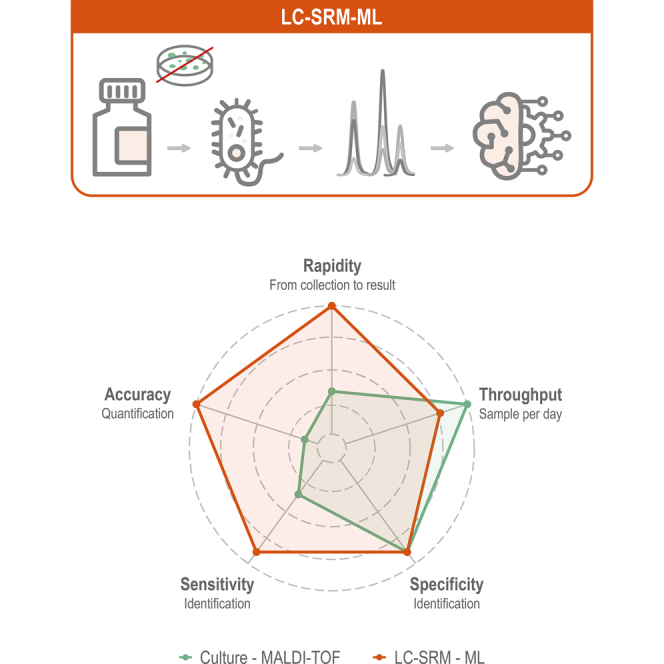
Urinary tract infections (UTIs) are a worldwide health problem. Fast and accurate detection of bacterial infection is essential to provide appropriate antibiotherapy to patients and to avoid the emergence of drug-resistant pathogens. While the gold standard requires 24 h to 48 h of bacteria culture prior to MALDI-TOF species identification, we propose a culture-free workflow, enabling bacterial identification and quantification in less than 4 h using 1 ml of urine. After rapid and automatable sample preparation, a signature of 82 bacterial peptides, defined by machine learning, was monitored in LC-MS, to distinguish the 15 species causing 84% of the UTIs. The combination of the sensitivity of the SRM mode on a triple quadrupole TSQ Altis instrument and the robustness of capillary flow enabled us to analyze up to 75 samples per day, with 99.2% accuracy on bacterial inoculations of healthy urines. We have also shown our method can be used to quantify the spread of the infection, from 8 × 104 to 3 × 107 CFU/ml. Finally, the workflow was validated on 45 inoculated urines and on 84 UTI-positive urine from patients, with respectively 93.3% and 87.1% of agreement with the culture-MALDI procedure at a level above 1 × 105 CFU/ml corresponding to an infection requiring antibiotherapy.
期刊介绍:
The mission of MCP is to foster the development and applications of proteomics in both basic and translational research. MCP will publish manuscripts that report significant new biological or clinical discoveries underpinned by proteomic observations across all kingdoms of life. Manuscripts must define the biological roles played by the proteins investigated or their mechanisms of action.
The journal also emphasizes articles that describe innovative new computational methods and technological advancements that will enable future discoveries. Manuscripts describing such approaches do not have to include a solution to a biological problem, but must demonstrate that the technology works as described, is reproducible and is appropriate to uncover yet unknown protein/proteome function or properties using relevant model systems or publicly available data.
Scope:
-Fundamental studies in biology, including integrative "omics" studies, that provide mechanistic insights
-Novel experimental and computational technologies
-Proteogenomic data integration and analysis that enable greater understanding of physiology and disease processes
-Pathway and network analyses of signaling that focus on the roles of post-translational modifications
-Studies of proteome dynamics and quality controls, and their roles in disease
-Studies of evolutionary processes effecting proteome dynamics, quality and regulation
-Chemical proteomics, including mechanisms of drug action
-Proteomics of the immune system and antigen presentation/recognition
-Microbiome proteomics, host-microbe and host-pathogen interactions, and their roles in health and disease
-Clinical and translational studies of human diseases
-Metabolomics to understand functional connections between genes, proteins and phenotypes

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


