{"title":"将库仑相互作用纳入通用神经网络势的液体电极界面分子动力学","authors":"Kaoru Hisama, Gerardo Valadez Huerta, Michihisa Koyama","doi":"10.1002/jcc.27487","DOIUrl":null,"url":null,"abstract":"<p>Computational understanding of the liquid–electrode interface faces challenges in efficiently incorporating reactive force fields and electrostatic potentials within reasonable computational costs. Although universal neural network potentials (UNNPs), representing pretrained machine learning interatomic potentials, are emerging, current UNNP models lack explicit treatment of Coulomb potentials, and methods for integrating additional charges on the electrode remain to be established. We propose a method to analyze liquid–electrode interfaces by integrating a UNNP, known as the preferred potential, with Coulomb potentials using the ONIOM method. This approach extends the applicability of UNNPs to electrode–liquid interface systems. Through molecular dynamics simulations of graphene–water and graphene oxide (GO)–water interfaces, we demonstrate the effectiveness of our method. Our findings emphasize the necessity of incorporating long-range Coulomb potentials into the water potential to accurately describe water polarization at the interface. Furthermore, we observe that functional groups on the GO electrode influence both polarization and capacitance.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2805-2811"},"PeriodicalIF":3.4000,"publicationDate":"2024-08-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27487","citationCount":"0","resultStr":"{\"title\":\"Molecular dynamics of liquid–electrode interface by integrating Coulomb interaction into universal neural network potential\",\"authors\":\"Kaoru Hisama, Gerardo Valadez Huerta, Michihisa Koyama\",\"doi\":\"10.1002/jcc.27487\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Computational understanding of the liquid–electrode interface faces challenges in efficiently incorporating reactive force fields and electrostatic potentials within reasonable computational costs. Although universal neural network potentials (UNNPs), representing pretrained machine learning interatomic potentials, are emerging, current UNNP models lack explicit treatment of Coulomb potentials, and methods for integrating additional charges on the electrode remain to be established. We propose a method to analyze liquid–electrode interfaces by integrating a UNNP, known as the preferred potential, with Coulomb potentials using the ONIOM method. This approach extends the applicability of UNNPs to electrode–liquid interface systems. Through molecular dynamics simulations of graphene–water and graphene oxide (GO)–water interfaces, we demonstrate the effectiveness of our method. Our findings emphasize the necessity of incorporating long-range Coulomb potentials into the water potential to accurately describe water polarization at the interface. Furthermore, we observe that functional groups on the GO electrode influence both polarization and capacitance.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 32\",\"pages\":\"2805-2811\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-08-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27487\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27487\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27487","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Molecular dynamics of liquid–electrode interface by integrating Coulomb interaction into universal neural network potential
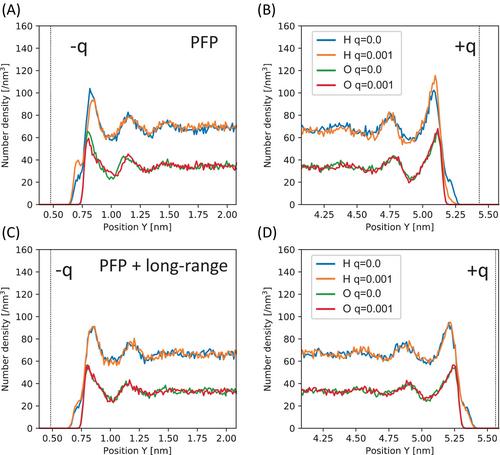
Computational understanding of the liquid–electrode interface faces challenges in efficiently incorporating reactive force fields and electrostatic potentials within reasonable computational costs. Although universal neural network potentials (UNNPs), representing pretrained machine learning interatomic potentials, are emerging, current UNNP models lack explicit treatment of Coulomb potentials, and methods for integrating additional charges on the electrode remain to be established. We propose a method to analyze liquid–electrode interfaces by integrating a UNNP, known as the preferred potential, with Coulomb potentials using the ONIOM method. This approach extends the applicability of UNNPs to electrode–liquid interface systems. Through molecular dynamics simulations of graphene–water and graphene oxide (GO)–water interfaces, we demonstrate the effectiveness of our method. Our findings emphasize the necessity of incorporating long-range Coulomb potentials into the water potential to accurately describe water polarization at the interface. Furthermore, we observe that functional groups on the GO electrode influence both polarization and capacitance.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


