Siyavash Moradi, Rebecca Tomann, Josie Hendrix, Martin Head-Gordon, Christopher J. Stein
{"title":"自旋极化扩展紧密结合方法的自旋参数优化。","authors":"Siyavash Moradi, Rebecca Tomann, Josie Hendrix, Martin Head-Gordon, Christopher J. Stein","doi":"10.1002/jcc.27482","DOIUrl":null,"url":null,"abstract":"<p>We present an optimization strategy for atom-specific spin-polarization constants within the spin-polarized GFN2-xTB framework, aiming to enhance the accuracy of molecular simulations. We compare a sequential and global optimization of spin parameters for hydrogen, carbon, nitrogen, oxygen, and fluorine. Sensitivity analysis using Sobol indices guides the identification of the most influential parameters for a given reference dataset, allowing for a nuanced understanding of their impact on diverse molecular properties. In the case of the W4-11 dataset, substantial error reduction was achieved, demonstrating the potential of the optimization. Transferability of the optimized spin-polarization constants over different properties, however, is limited, as we demonstrate by applying the optimized parameters on a set of singlet-triplet gaps in carbenes. Further studies on ionization potentials and electron affinities highlight some inherent limitations of current extended tight-binding methods that can not be resolved by simple parameter optimization. We conclude that the significantly improved accuracy strongly encourages the present re-optimization of the spin-polarization constants, whereas the limited transferability motivates a property-specific optimization strategy.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2786-2792"},"PeriodicalIF":3.4000,"publicationDate":"2024-08-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27482","citationCount":"0","resultStr":"{\"title\":\"Spin parameter optimization for spin-polarized extended tight-binding methods\",\"authors\":\"Siyavash Moradi, Rebecca Tomann, Josie Hendrix, Martin Head-Gordon, Christopher J. Stein\",\"doi\":\"10.1002/jcc.27482\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>We present an optimization strategy for atom-specific spin-polarization constants within the spin-polarized GFN2-xTB framework, aiming to enhance the accuracy of molecular simulations. We compare a sequential and global optimization of spin parameters for hydrogen, carbon, nitrogen, oxygen, and fluorine. Sensitivity analysis using Sobol indices guides the identification of the most influential parameters for a given reference dataset, allowing for a nuanced understanding of their impact on diverse molecular properties. In the case of the W4-11 dataset, substantial error reduction was achieved, demonstrating the potential of the optimization. Transferability of the optimized spin-polarization constants over different properties, however, is limited, as we demonstrate by applying the optimized parameters on a set of singlet-triplet gaps in carbenes. Further studies on ionization potentials and electron affinities highlight some inherent limitations of current extended tight-binding methods that can not be resolved by simple parameter optimization. We conclude that the significantly improved accuracy strongly encourages the present re-optimization of the spin-polarization constants, whereas the limited transferability motivates a property-specific optimization strategy.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 32\",\"pages\":\"2786-2792\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-08-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27482\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27482\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27482","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Spin parameter optimization for spin-polarized extended tight-binding methods
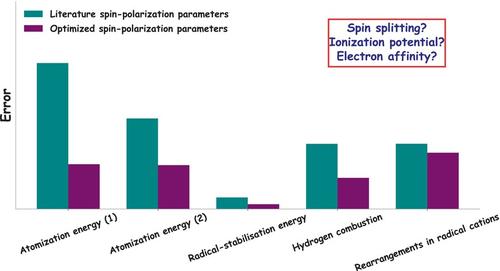
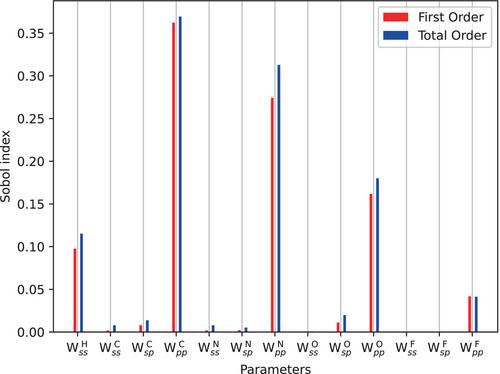
We present an optimization strategy for atom-specific spin-polarization constants within the spin-polarized GFN2-xTB framework, aiming to enhance the accuracy of molecular simulations. We compare a sequential and global optimization of spin parameters for hydrogen, carbon, nitrogen, oxygen, and fluorine. Sensitivity analysis using Sobol indices guides the identification of the most influential parameters for a given reference dataset, allowing for a nuanced understanding of their impact on diverse molecular properties. In the case of the W4-11 dataset, substantial error reduction was achieved, demonstrating the potential of the optimization. Transferability of the optimized spin-polarization constants over different properties, however, is limited, as we demonstrate by applying the optimized parameters on a set of singlet-triplet gaps in carbenes. Further studies on ionization potentials and electron affinities highlight some inherent limitations of current extended tight-binding methods that can not be resolved by simple parameter optimization. We conclude that the significantly improved accuracy strongly encourages the present re-optimization of the spin-polarization constants, whereas the limited transferability motivates a property-specific optimization strategy.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


