Rosita Del Prete, Roberta Drago, Federica Nardi, Gaia Bartolini, Erika Bellini, Antonella De Rosa, Silvia Valensin, Anna Kabanova
{"title":"在 Eµ-TCL1 慢性淋巴细胞白血病模型中对原发肿瘤 B 细胞进行可靠且经济高效的 CRISPR/Cas9 基因编辑","authors":"Rosita Del Prete, Roberta Drago, Federica Nardi, Gaia Bartolini, Erika Bellini, Antonella De Rosa, Silvia Valensin, Anna Kabanova","doi":"10.1002/hem3.134","DOIUrl":null,"url":null,"abstract":"<p>Ability to genetically edit primary B cells via CRISPR/Cas9 technology represents a powerful tool to study molecular mechanisms of B-cell pathogenesis. In this context, employing ribonucleoprotein complexes (RNPs), formed by recombinant Cas9 and genome-targetting single guide RNA molecules, brings in advantage of accelerated set-up and protocol robustness. Gene editing via RNP electroporation has been recently applied to primary tumor cells isolated from patients chronic lymphocytic leukemia (CLL), suggesting an efficient and valuable tool for studying leukemic cell biology and biomarker validation.<span><sup>1, 2</sup></span> The work by Nardi et al. on this topic proposed to electroporate unmanipulated primary CLL cells that are subsequently put in culture with human CD40L-expressing fibroblasts and soluble stimuli to promote CLL cell proliferation. In this context, cellular proliferation is required to achieve homozygous gene editing, whereas in unstimulated CLL cells it is possible to achieve only the heterozygous editing.<span><sup>1</sup></span> The method published by Mateos-Jaimez et al. relies on the preactivation of CLL cells with CD40L/BAFF/IL-21-expressing stromal cells, followed by RNP electroporation and continuation of the stimulatory coculture.<span><sup>2</sup></span> Both methods approach 80%–90% of editing efficiency and allow to perform downstream <i>in vitro</i> experiments on edited leukemic cells.</p><p>Application of a similar RNP-based editing approach to the widely used murine model of CLL, the Eμ-TCL1 transgenic mice,<span><sup>3</sup></span> represents a valuable and versatile tool to explore CLL biology <i>in vivo</i>. Examples illustrating its feasibility has been first shown in studies by Chakraborthy et al. and Martines et al.<span><sup>4, 5</sup></span> The published method consists in preactivating primary CD19<sup>+</sup>CD5<sup>+</sup> leukemic B cells by TLR9 agonist CpG ODN-1668, followed by RNP electroporation and intraperitoneal injection of 30 × 10<sup>6</sup> electroporated cells to promote expansion of edited leukemic cells <i>in vivo</i>. Despite this method has been proven effective, it has not been set up to expand edited TCL1 cells <i>in vitro</i>. This is associated with high experimental costs and does not allow to perform functional analysis of gene editing phenotype prior to the <i>in vivo</i> transfer, which eventually becomes not feasible if edited cells are unfit <i>in vivo</i>.</p><p>Hence, we envisioned a new approach that would allow to expand RNP-electroporated TCL1 cells <i>in vitro</i> prior to transfer. To this end, we first optimized culture conditions for TCL1 cells evaluating their viability and proliferation after treatment with different stimuli. We observed that ODN-1668 stimulation, although being efficient in activating TCL1 cells in the short-term,<span><sup>5</sup></span> does not allow to expand them <i>in vitro</i> (Supporting Information S1: Figure S1). We thus evaluated different stimulation conditions and established that coculture of TCL1 cells with irradiated fibroblasts expressing murine CD40L was sufficient to induce proliferation of leukemic cells over 5 days of culture (Figure 1A,B). Curiously, IL-4 and IL-21 addition did not enhance CD40-driven TCL1 cell growth, in contrast to what is typically observed for human CLL cells (Figure 1A).</p><p>Next, we optimized electroporation settings for TCL1 cells using an <i>in vitro</i> translated mRNA encoding GFP<span><sup>1</sup></span> and an electroporation system with bimodal pulsing strategy previously employed by us for human CLL cell electroporation.<span><sup>1</sup></span> Among 13 tested electroporation conditions, the optimal one allowed us to maintain >90% vitality of recovered TCL1 cells and achieve around 80% cell transfection efficiency (Figure 1C; electroporation settings in Supporting Information S1: Table S1; gating strategy in Supporting Information S1: Figure S2). To set up a protocol for CRISPR/Cas9 gene editing of TCL1 cells, we followed a pipeline indicated in Figure 1D. Electroporating TCL1 cells with polyglutamic acid-stabilized RNPs<span><sup>7</sup></span> and allowing them to proliferate 4–5 days post-electroporation typically allowed us to achieve >80% gene KO in bulk population, either with single (Figure 1E,F) or double combination of sgRNAs (Figure 1G,H and Supporting Information S1: Figure S3). For example, targeting the surface marker CD19 proved convenient as a control that could be easily assessed by flow cytometry (Figure 1G,H). The protocol typically allowed for three-fold expansion of electroporated TCL1 cells, while maximum levels of gene silencing could be achieved by Day 4 of culturing (Figure S4). To expand Cas9/sgRNA-electroporated TCL1 cells <i>in vivo</i>, we used intravenous (<i>i.v.</i>) route of tumor infection into healthy C57BL/6 recipients<span><sup>8</sup></span> allowing for homogeneous tumor engraftment in C57BL/6 recipients at a reduced dose (5 × 10<sup>6</sup> cells per animal; Supporting Information S1: Figure S5).</p><p>In order to apply the newly developed protocol (Supporting Information Methods) to validate a relevant biomarker implicated in CLL pathogenesis, we decided to target the costimulatory receptor CD40, the triggering of which is sufficient to induce TCL1 cell proliferation (Figure 1A). CD40 engagement within CLL proliferative centers is thought to dictate CLL progression <i>in vivo</i><span><sup>9</sup></span> and promote resistance to therapies such as the Bcl2 inhibitor venetoclax.<span><sup>10</sup></span> We established sgRNA combination that was highly efficient in <i>Cd40</i> silencing <i>in vitro</i>, while silencing <i>Cd4</i> which is not expected to give any phenotype in TCL1 cells as a control (Figure 2A,B). Contrary to what expected, we observed that <i>Cd40</i> silencing did not affect CD40-driven TCL1 cell proliferation <i>in vitro</i>, suggesting that initial levels of CD40 protein might be sufficient to prime cells for proliferation and that at later time points CD40 signaling becomes dispensable (Figure 2C). We then performed an <i>in vivo</i> competition experiment by injecting <i>Cd40</i>-edited and <i>Cd4</i>-edited TCL1 cells into C57BL/6 recipients (Figure 2D). Resulting tumors maintained constant levels of <i>Cd40</i>-silenced tumor population that persisted <i>in vivo</i>, which was reflected in the constant frequency of <i>Cd40</i> locus indels in bulk tumor population (Figure 2D, right panel) and in a significantly lower CD40 expression compared to control tumors originating from <i>Cd4</i>-edited cells (Figure 2E,F). Our findings hence suggested that the loss of CD40 expression did not compromise expansion of leukemic cells <i>in vivo</i>. This result goes in line with a recently published observation that wild-type TCL1 cells are able to expand in CD40L<sup>−/−</sup> hosts.<span><sup>11</sup></span> Hence, complementary approaches allowing to test gene function in leukemia progression both by disrupting its expression on tumor cells, or within tumor microenvironment,<span><sup>11</sup></span> confidently illustrates that CD40 function may be substituted by other proliferative stimuli within CLL niche <i>in vivo</i>.</p><p>Our work reports a highly efficient and easily controlled genetic modification protocol for primary leukemic B cells in the TCL1 model. The whole cycle of gene silencing could be completed within 6–8 weeks from the start of sgRNA validation step, allowing to rapidly evaluate gene function in CLL progression. Our protocol routinely achieves over 80% editing efficiency and allows for quality control of CRISPR/Cas9 editing and functional assessment of gene silencing already in the <i>in vitro</i> phase. It lowers the cost of editing experiments as the <i>in vitro</i> TCL1 cell expansion allows for up to three-fold multiplication of edited cell population and proves effective in engrafting as low as 5 × 10<sup>6</sup> of edited TCL1 cells per animal via <i>i.v.</i> administration. Finally, our method is applicable to multiplexed gene editing <i>in vitro</i> and <i>in vivo</i> (Supporting Information S1: Figure S6) and could be translated to precise genome editing, wherein Cas9 RNPs are co-electroporated with a DNA template for homology-directed repair,<span><sup>12-15</sup></span> and to other B cell types, after appropriate set up of electroporation and expansion conditions.</p><p>Rosita Del Prete and Roberta Drago conceived the experimental design, performed experiments, analysed data and wrote the manuscript; Rosita Del Prete, Roberta Drago, Federica Nardi, Gaia Bartolini, Erika Bellini, Antonella De Rosa, and Silvia Valensin performed and analyzed experiments; Anna Kabanova coordinated the project, supervised experimental design, consulted obtained results, edited the manuscript and provided funding.</p><p>The authors declare no conflict of interest.</p>","PeriodicalId":12982,"journal":{"name":"HemaSphere","volume":"8 8","pages":""},"PeriodicalIF":7.6000,"publicationDate":"2024-08-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/hem3.134","citationCount":"0","resultStr":"{\"title\":\"Robust and cost-effective CRISPR/Cas9 gene editing of primary tumor B cells in Eµ-TCL1 model of chronic lymphocytic leukemia\",\"authors\":\"Rosita Del Prete, Roberta Drago, Federica Nardi, Gaia Bartolini, Erika Bellini, Antonella De Rosa, Silvia Valensin, Anna Kabanova\",\"doi\":\"10.1002/hem3.134\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Ability to genetically edit primary B cells via CRISPR/Cas9 technology represents a powerful tool to study molecular mechanisms of B-cell pathogenesis. In this context, employing ribonucleoprotein complexes (RNPs), formed by recombinant Cas9 and genome-targetting single guide RNA molecules, brings in advantage of accelerated set-up and protocol robustness. Gene editing via RNP electroporation has been recently applied to primary tumor cells isolated from patients chronic lymphocytic leukemia (CLL), suggesting an efficient and valuable tool for studying leukemic cell biology and biomarker validation.<span><sup>1, 2</sup></span> The work by Nardi et al. on this topic proposed to electroporate unmanipulated primary CLL cells that are subsequently put in culture with human CD40L-expressing fibroblasts and soluble stimuli to promote CLL cell proliferation. In this context, cellular proliferation is required to achieve homozygous gene editing, whereas in unstimulated CLL cells it is possible to achieve only the heterozygous editing.<span><sup>1</sup></span> The method published by Mateos-Jaimez et al. relies on the preactivation of CLL cells with CD40L/BAFF/IL-21-expressing stromal cells, followed by RNP electroporation and continuation of the stimulatory coculture.<span><sup>2</sup></span> Both methods approach 80%–90% of editing efficiency and allow to perform downstream <i>in vitro</i> experiments on edited leukemic cells.</p><p>Application of a similar RNP-based editing approach to the widely used murine model of CLL, the Eμ-TCL1 transgenic mice,<span><sup>3</sup></span> represents a valuable and versatile tool to explore CLL biology <i>in vivo</i>. Examples illustrating its feasibility has been first shown in studies by Chakraborthy et al. and Martines et al.<span><sup>4, 5</sup></span> The published method consists in preactivating primary CD19<sup>+</sup>CD5<sup>+</sup> leukemic B cells by TLR9 agonist CpG ODN-1668, followed by RNP electroporation and intraperitoneal injection of 30 × 10<sup>6</sup> electroporated cells to promote expansion of edited leukemic cells <i>in vivo</i>. Despite this method has been proven effective, it has not been set up to expand edited TCL1 cells <i>in vitro</i>. This is associated with high experimental costs and does not allow to perform functional analysis of gene editing phenotype prior to the <i>in vivo</i> transfer, which eventually becomes not feasible if edited cells are unfit <i>in vivo</i>.</p><p>Hence, we envisioned a new approach that would allow to expand RNP-electroporated TCL1 cells <i>in vitro</i> prior to transfer. To this end, we first optimized culture conditions for TCL1 cells evaluating their viability and proliferation after treatment with different stimuli. We observed that ODN-1668 stimulation, although being efficient in activating TCL1 cells in the short-term,<span><sup>5</sup></span> does not allow to expand them <i>in vitro</i> (Supporting Information S1: Figure S1). We thus evaluated different stimulation conditions and established that coculture of TCL1 cells with irradiated fibroblasts expressing murine CD40L was sufficient to induce proliferation of leukemic cells over 5 days of culture (Figure 1A,B). Curiously, IL-4 and IL-21 addition did not enhance CD40-driven TCL1 cell growth, in contrast to what is typically observed for human CLL cells (Figure 1A).</p><p>Next, we optimized electroporation settings for TCL1 cells using an <i>in vitro</i> translated mRNA encoding GFP<span><sup>1</sup></span> and an electroporation system with bimodal pulsing strategy previously employed by us for human CLL cell electroporation.<span><sup>1</sup></span> Among 13 tested electroporation conditions, the optimal one allowed us to maintain >90% vitality of recovered TCL1 cells and achieve around 80% cell transfection efficiency (Figure 1C; electroporation settings in Supporting Information S1: Table S1; gating strategy in Supporting Information S1: Figure S2). To set up a protocol for CRISPR/Cas9 gene editing of TCL1 cells, we followed a pipeline indicated in Figure 1D. Electroporating TCL1 cells with polyglutamic acid-stabilized RNPs<span><sup>7</sup></span> and allowing them to proliferate 4–5 days post-electroporation typically allowed us to achieve >80% gene KO in bulk population, either with single (Figure 1E,F) or double combination of sgRNAs (Figure 1G,H and Supporting Information S1: Figure S3). For example, targeting the surface marker CD19 proved convenient as a control that could be easily assessed by flow cytometry (Figure 1G,H). The protocol typically allowed for three-fold expansion of electroporated TCL1 cells, while maximum levels of gene silencing could be achieved by Day 4 of culturing (Figure S4). To expand Cas9/sgRNA-electroporated TCL1 cells <i>in vivo</i>, we used intravenous (<i>i.v.</i>) route of tumor infection into healthy C57BL/6 recipients<span><sup>8</sup></span> allowing for homogeneous tumor engraftment in C57BL/6 recipients at a reduced dose (5 × 10<sup>6</sup> cells per animal; Supporting Information S1: Figure S5).</p><p>In order to apply the newly developed protocol (Supporting Information Methods) to validate a relevant biomarker implicated in CLL pathogenesis, we decided to target the costimulatory receptor CD40, the triggering of which is sufficient to induce TCL1 cell proliferation (Figure 1A). CD40 engagement within CLL proliferative centers is thought to dictate CLL progression <i>in vivo</i><span><sup>9</sup></span> and promote resistance to therapies such as the Bcl2 inhibitor venetoclax.<span><sup>10</sup></span> We established sgRNA combination that was highly efficient in <i>Cd40</i> silencing <i>in vitro</i>, while silencing <i>Cd4</i> which is not expected to give any phenotype in TCL1 cells as a control (Figure 2A,B). Contrary to what expected, we observed that <i>Cd40</i> silencing did not affect CD40-driven TCL1 cell proliferation <i>in vitro</i>, suggesting that initial levels of CD40 protein might be sufficient to prime cells for proliferation and that at later time points CD40 signaling becomes dispensable (Figure 2C). We then performed an <i>in vivo</i> competition experiment by injecting <i>Cd40</i>-edited and <i>Cd4</i>-edited TCL1 cells into C57BL/6 recipients (Figure 2D). Resulting tumors maintained constant levels of <i>Cd40</i>-silenced tumor population that persisted <i>in vivo</i>, which was reflected in the constant frequency of <i>Cd40</i> locus indels in bulk tumor population (Figure 2D, right panel) and in a significantly lower CD40 expression compared to control tumors originating from <i>Cd4</i>-edited cells (Figure 2E,F). Our findings hence suggested that the loss of CD40 expression did not compromise expansion of leukemic cells <i>in vivo</i>. This result goes in line with a recently published observation that wild-type TCL1 cells are able to expand in CD40L<sup>−/−</sup> hosts.<span><sup>11</sup></span> Hence, complementary approaches allowing to test gene function in leukemia progression both by disrupting its expression on tumor cells, or within tumor microenvironment,<span><sup>11</sup></span> confidently illustrates that CD40 function may be substituted by other proliferative stimuli within CLL niche <i>in vivo</i>.</p><p>Our work reports a highly efficient and easily controlled genetic modification protocol for primary leukemic B cells in the TCL1 model. The whole cycle of gene silencing could be completed within 6–8 weeks from the start of sgRNA validation step, allowing to rapidly evaluate gene function in CLL progression. Our protocol routinely achieves over 80% editing efficiency and allows for quality control of CRISPR/Cas9 editing and functional assessment of gene silencing already in the <i>in vitro</i> phase. It lowers the cost of editing experiments as the <i>in vitro</i> TCL1 cell expansion allows for up to three-fold multiplication of edited cell population and proves effective in engrafting as low as 5 × 10<sup>6</sup> of edited TCL1 cells per animal via <i>i.v.</i> administration. Finally, our method is applicable to multiplexed gene editing <i>in vitro</i> and <i>in vivo</i> (Supporting Information S1: Figure S6) and could be translated to precise genome editing, wherein Cas9 RNPs are co-electroporated with a DNA template for homology-directed repair,<span><sup>12-15</sup></span> and to other B cell types, after appropriate set up of electroporation and expansion conditions.</p><p>Rosita Del Prete and Roberta Drago conceived the experimental design, performed experiments, analysed data and wrote the manuscript; Rosita Del Prete, Roberta Drago, Federica Nardi, Gaia Bartolini, Erika Bellini, Antonella De Rosa, and Silvia Valensin performed and analyzed experiments; Anna Kabanova coordinated the project, supervised experimental design, consulted obtained results, edited the manuscript and provided funding.</p><p>The authors declare no conflict of interest.</p>\",\"PeriodicalId\":12982,\"journal\":{\"name\":\"HemaSphere\",\"volume\":\"8 8\",\"pages\":\"\"},\"PeriodicalIF\":7.6000,\"publicationDate\":\"2024-08-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/hem3.134\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"HemaSphere\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/hem3.134\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"HemaSphere","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/hem3.134","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
Robust and cost-effective CRISPR/Cas9 gene editing of primary tumor B cells in Eµ-TCL1 model of chronic lymphocytic leukemia
Ability to genetically edit primary B cells via CRISPR/Cas9 technology represents a powerful tool to study molecular mechanisms of B-cell pathogenesis. In this context, employing ribonucleoprotein complexes (RNPs), formed by recombinant Cas9 and genome-targetting single guide RNA molecules, brings in advantage of accelerated set-up and protocol robustness. Gene editing via RNP electroporation has been recently applied to primary tumor cells isolated from patients chronic lymphocytic leukemia (CLL), suggesting an efficient and valuable tool for studying leukemic cell biology and biomarker validation.1, 2 The work by Nardi et al. on this topic proposed to electroporate unmanipulated primary CLL cells that are subsequently put in culture with human CD40L-expressing fibroblasts and soluble stimuli to promote CLL cell proliferation. In this context, cellular proliferation is required to achieve homozygous gene editing, whereas in unstimulated CLL cells it is possible to achieve only the heterozygous editing.1 The method published by Mateos-Jaimez et al. relies on the preactivation of CLL cells with CD40L/BAFF/IL-21-expressing stromal cells, followed by RNP electroporation and continuation of the stimulatory coculture.2 Both methods approach 80%–90% of editing efficiency and allow to perform downstream in vitro experiments on edited leukemic cells.
Application of a similar RNP-based editing approach to the widely used murine model of CLL, the Eμ-TCL1 transgenic mice,3 represents a valuable and versatile tool to explore CLL biology in vivo. Examples illustrating its feasibility has been first shown in studies by Chakraborthy et al. and Martines et al.4, 5 The published method consists in preactivating primary CD19+CD5+ leukemic B cells by TLR9 agonist CpG ODN-1668, followed by RNP electroporation and intraperitoneal injection of 30 × 106 electroporated cells to promote expansion of edited leukemic cells in vivo. Despite this method has been proven effective, it has not been set up to expand edited TCL1 cells in vitro. This is associated with high experimental costs and does not allow to perform functional analysis of gene editing phenotype prior to the in vivo transfer, which eventually becomes not feasible if edited cells are unfit in vivo.
Hence, we envisioned a new approach that would allow to expand RNP-electroporated TCL1 cells in vitro prior to transfer. To this end, we first optimized culture conditions for TCL1 cells evaluating their viability and proliferation after treatment with different stimuli. We observed that ODN-1668 stimulation, although being efficient in activating TCL1 cells in the short-term,5 does not allow to expand them in vitro (Supporting Information S1: Figure S1). We thus evaluated different stimulation conditions and established that coculture of TCL1 cells with irradiated fibroblasts expressing murine CD40L was sufficient to induce proliferation of leukemic cells over 5 days of culture (Figure 1A,B). Curiously, IL-4 and IL-21 addition did not enhance CD40-driven TCL1 cell growth, in contrast to what is typically observed for human CLL cells (Figure 1A).
Next, we optimized electroporation settings for TCL1 cells using an in vitro translated mRNA encoding GFP1 and an electroporation system with bimodal pulsing strategy previously employed by us for human CLL cell electroporation.1 Among 13 tested electroporation conditions, the optimal one allowed us to maintain >90% vitality of recovered TCL1 cells and achieve around 80% cell transfection efficiency (Figure 1C; electroporation settings in Supporting Information S1: Table S1; gating strategy in Supporting Information S1: Figure S2). To set up a protocol for CRISPR/Cas9 gene editing of TCL1 cells, we followed a pipeline indicated in Figure 1D. Electroporating TCL1 cells with polyglutamic acid-stabilized RNPs7 and allowing them to proliferate 4–5 days post-electroporation typically allowed us to achieve >80% gene KO in bulk population, either with single (Figure 1E,F) or double combination of sgRNAs (Figure 1G,H and Supporting Information S1: Figure S3). For example, targeting the surface marker CD19 proved convenient as a control that could be easily assessed by flow cytometry (Figure 1G,H). The protocol typically allowed for three-fold expansion of electroporated TCL1 cells, while maximum levels of gene silencing could be achieved by Day 4 of culturing (Figure S4). To expand Cas9/sgRNA-electroporated TCL1 cells in vivo, we used intravenous (i.v.) route of tumor infection into healthy C57BL/6 recipients8 allowing for homogeneous tumor engraftment in C57BL/6 recipients at a reduced dose (5 × 106 cells per animal; Supporting Information S1: Figure S5).
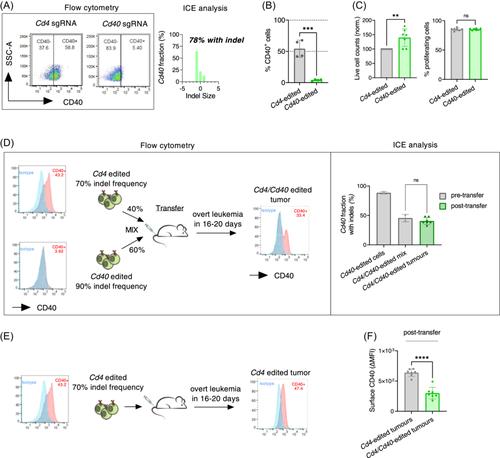
In order to apply the newly developed protocol (Supporting Information Methods) to validate a relevant biomarker implicated in CLL pathogenesis, we decided to target the costimulatory receptor CD40, the triggering of which is sufficient to induce TCL1 cell proliferation (Figure 1A). CD40 engagement within CLL proliferative centers is thought to dictate CLL progression in vivo9 and promote resistance to therapies such as the Bcl2 inhibitor venetoclax.10 We established sgRNA combination that was highly efficient in Cd40 silencing in vitro, while silencing Cd4 which is not expected to give any phenotype in TCL1 cells as a control (Figure 2A,B). Contrary to what expected, we observed that Cd40 silencing did not affect CD40-driven TCL1 cell proliferation in vitro, suggesting that initial levels of CD40 protein might be sufficient to prime cells for proliferation and that at later time points CD40 signaling becomes dispensable (Figure 2C). We then performed an in vivo competition experiment by injecting Cd40-edited and Cd4-edited TCL1 cells into C57BL/6 recipients (Figure 2D). Resulting tumors maintained constant levels of Cd40-silenced tumor population that persisted in vivo, which was reflected in the constant frequency of Cd40 locus indels in bulk tumor population (Figure 2D, right panel) and in a significantly lower CD40 expression compared to control tumors originating from Cd4-edited cells (Figure 2E,F). Our findings hence suggested that the loss of CD40 expression did not compromise expansion of leukemic cells in vivo. This result goes in line with a recently published observation that wild-type TCL1 cells are able to expand in CD40L−/− hosts.11 Hence, complementary approaches allowing to test gene function in leukemia progression both by disrupting its expression on tumor cells, or within tumor microenvironment,11 confidently illustrates that CD40 function may be substituted by other proliferative stimuli within CLL niche in vivo.
Our work reports a highly efficient and easily controlled genetic modification protocol for primary leukemic B cells in the TCL1 model. The whole cycle of gene silencing could be completed within 6–8 weeks from the start of sgRNA validation step, allowing to rapidly evaluate gene function in CLL progression. Our protocol routinely achieves over 80% editing efficiency and allows for quality control of CRISPR/Cas9 editing and functional assessment of gene silencing already in the in vitro phase. It lowers the cost of editing experiments as the in vitro TCL1 cell expansion allows for up to three-fold multiplication of edited cell population and proves effective in engrafting as low as 5 × 106 of edited TCL1 cells per animal via i.v. administration. Finally, our method is applicable to multiplexed gene editing in vitro and in vivo (Supporting Information S1: Figure S6) and could be translated to precise genome editing, wherein Cas9 RNPs are co-electroporated with a DNA template for homology-directed repair,12-15 and to other B cell types, after appropriate set up of electroporation and expansion conditions.
Rosita Del Prete and Roberta Drago conceived the experimental design, performed experiments, analysed data and wrote the manuscript; Rosita Del Prete, Roberta Drago, Federica Nardi, Gaia Bartolini, Erika Bellini, Antonella De Rosa, and Silvia Valensin performed and analyzed experiments; Anna Kabanova coordinated the project, supervised experimental design, consulted obtained results, edited the manuscript and provided funding.
期刊介绍:
HemaSphere, as a publication, is dedicated to disseminating the outcomes of profoundly pertinent basic, translational, and clinical research endeavors within the field of hematology. The journal actively seeks robust studies that unveil novel discoveries with significant ramifications for hematology.
In addition to original research, HemaSphere features review articles and guideline articles that furnish lucid synopses and discussions of emerging developments, along with recommendations for patient care.
Positioned as the foremost resource in hematology, HemaSphere augments its offerings with specialized sections like HemaTopics and HemaPolicy. These segments engender insightful dialogues covering a spectrum of hematology-related topics, including digestible summaries of pivotal articles, updates on new therapies, deliberations on European policy matters, and other noteworthy news items within the field. Steering the course of HemaSphere are Editor in Chief Jan Cools and Deputy Editor in Chief Claire Harrison, alongside the guidance of an esteemed Editorial Board comprising international luminaries in both research and clinical realms, each representing diverse areas of hematologic expertise.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


