Jerimiah A. Zamora, Armando de Rezende, Reed Nieman, Neil Vaz, Andrew R. Demko, Michelle L. Pantoya, Daniel Tunega, Adelia J. A. Aquino
{"title":"金红石和锐钛矿表面的高氯酸铵吸附反应模型。","authors":"Jerimiah A. Zamora, Armando de Rezende, Reed Nieman, Neil Vaz, Andrew R. Demko, Michelle L. Pantoya, Daniel Tunega, Adelia J. A. Aquino","doi":"10.1002/jcc.27476","DOIUrl":null,"url":null,"abstract":"<p>In this work, the effects of two TiO<sub>2</sub> polymorphs on the decomposition of ammonium perchlorate (NH<sub>4</sub>ClO<sub>4</sub>) were studied experimentally and theoretically. The interactions between AP and various surfaces of TiO<sub>2</sub> were modeled using density functional theory (DFT) calculations. Specifically, the adsorption of AP on three rutile surfaces (1 1 0), (1 0 0), and (0 0 1), as well as two anatase surfaces (1 0 1), and (0 0 1) were modeled using cluster models, along with the decomposition of adsorbed AP into small molecules. The optimized complexes of the AP molecule on TiO<sub>2</sub> surfaces were very stable, indicating strong covalent and hydrogen bonding interactions, leading to highly energetic adsorption reactions. The calculated energy of adsorption (Δ<i>E</i><sub>ads</sub>) ranged from −120.23 to −301.98 kJ/mol, with highly exergonic calculated Gibbs free energy (Δ<i>G</i><sub>ads</sub>) of reaction, and highly exothermic enthalpy of reaction (Δ<i>H</i><sub>ads</sub>). The decomposition of adsorbed AP was also found to have very negative Δ<i>E</i><sub>dec</sub> values between −199.08 and −380.73 kJ/mol. The values of Δ<i>G</i><sub>dec</sub> and Δ<i>H</i><sub>dec</sub> reveal exergonic and exothermic reactions. The adsorption of AP on TiO<sub>2</sub> surfaces anticipates the heat release of decomposition, in agreement with experimental results. The most common anatase surface, (1 0 1), was predicted to be more reactive for AP decomposition than the most stable rutile surface, (1 1 0), which was confirmed by experiments. DFT calculations show the mechanism for activation of the two TiO<sub>2</sub> polymorphs is entropy driven.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2739-2748"},"PeriodicalIF":3.4000,"publicationDate":"2024-08-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Modeling adsorption reactions of ammonium perchlorate on rutile and anatase surfaces\",\"authors\":\"Jerimiah A. Zamora, Armando de Rezende, Reed Nieman, Neil Vaz, Andrew R. Demko, Michelle L. Pantoya, Daniel Tunega, Adelia J. A. Aquino\",\"doi\":\"10.1002/jcc.27476\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>In this work, the effects of two TiO<sub>2</sub> polymorphs on the decomposition of ammonium perchlorate (NH<sub>4</sub>ClO<sub>4</sub>) were studied experimentally and theoretically. The interactions between AP and various surfaces of TiO<sub>2</sub> were modeled using density functional theory (DFT) calculations. Specifically, the adsorption of AP on three rutile surfaces (1 1 0), (1 0 0), and (0 0 1), as well as two anatase surfaces (1 0 1), and (0 0 1) were modeled using cluster models, along with the decomposition of adsorbed AP into small molecules. The optimized complexes of the AP molecule on TiO<sub>2</sub> surfaces were very stable, indicating strong covalent and hydrogen bonding interactions, leading to highly energetic adsorption reactions. The calculated energy of adsorption (Δ<i>E</i><sub>ads</sub>) ranged from −120.23 to −301.98 kJ/mol, with highly exergonic calculated Gibbs free energy (Δ<i>G</i><sub>ads</sub>) of reaction, and highly exothermic enthalpy of reaction (Δ<i>H</i><sub>ads</sub>). The decomposition of adsorbed AP was also found to have very negative Δ<i>E</i><sub>dec</sub> values between −199.08 and −380.73 kJ/mol. The values of Δ<i>G</i><sub>dec</sub> and Δ<i>H</i><sub>dec</sub> reveal exergonic and exothermic reactions. The adsorption of AP on TiO<sub>2</sub> surfaces anticipates the heat release of decomposition, in agreement with experimental results. The most common anatase surface, (1 0 1), was predicted to be more reactive for AP decomposition than the most stable rutile surface, (1 1 0), which was confirmed by experiments. DFT calculations show the mechanism for activation of the two TiO<sub>2</sub> polymorphs is entropy driven.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 32\",\"pages\":\"2739-2748\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-08-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27476\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27476","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
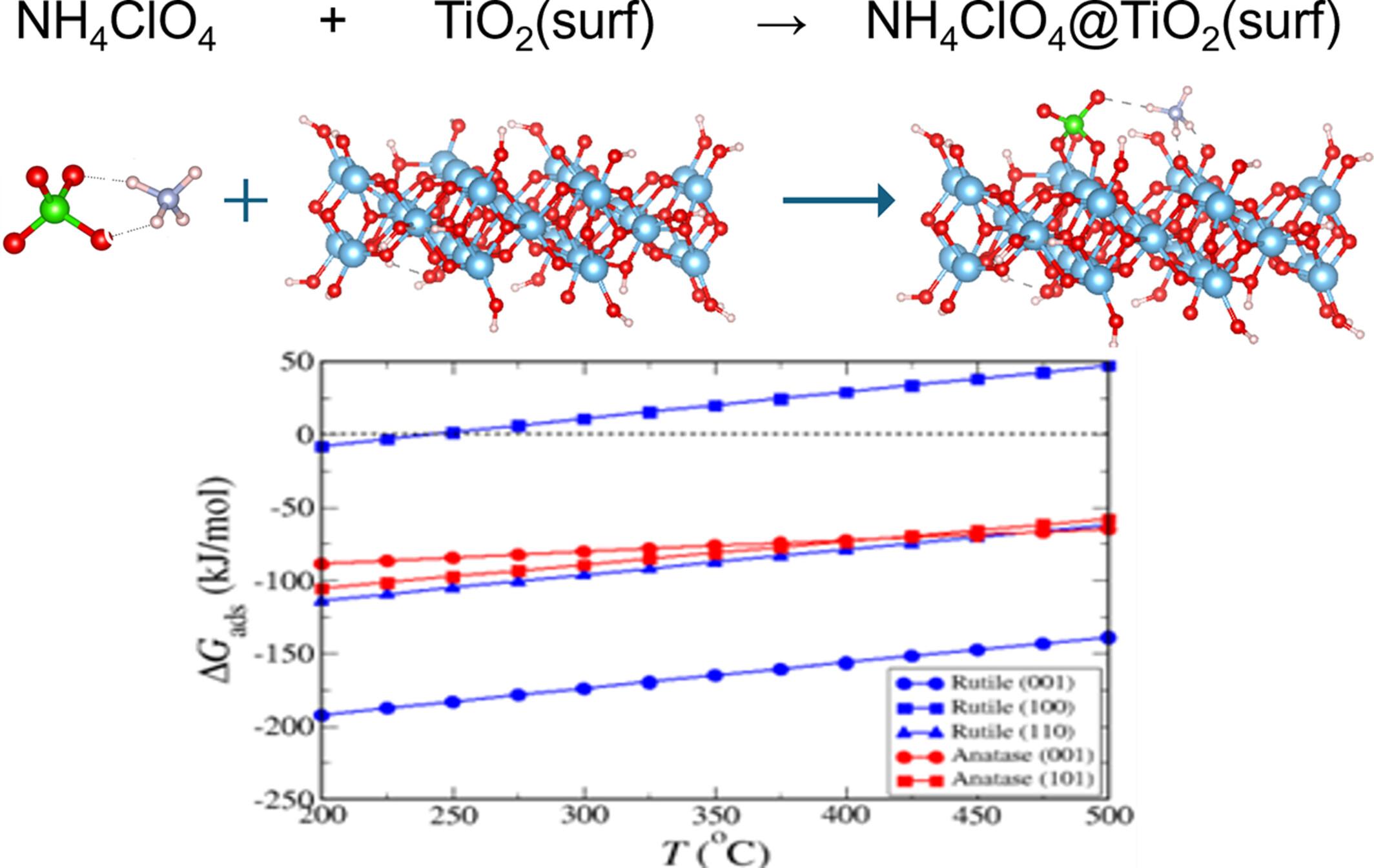
本研究从实验和理论两方面研究了两种二氧化钛多晶体对高氯酸铵(NH4ClO4)分解的影响。利用密度泛函理论(DFT)计算建立了 AP 与 TiO2 不同表面之间的相互作用模型。具体来说,利用簇模型对 AP 在三个金红石表面(1 1 0)、(1 0 0)和(0 0 1)以及两个锐钛矿表面(1 0 1)和(0 0 1)上的吸附进行了建模,同时对吸附的 AP 分解成小分子进行了建模。钛白粉分子在二氧化钛表面上的优化复合物非常稳定,表明共价键和氢键相互作用很强,导致了高能吸附反应。计算得出的吸附能(ΔEads)范围为-120.23 至 -301.98 kJ/mol,计算得出的反应吉布斯自由能(ΔGads)为高放热反应,反应焓(ΔHads)为高放热反应。还发现吸附 AP 的分解具有非常负的 ΔEdec 值,介于 -199.08 和 -380.73 kJ/mol 之间。ΔGdec和ΔHdec值显示了放热反应和放热反应。AP 在二氧化钛表面的吸附预示着分解热量的释放,这与实验结果一致。根据预测,最常见的锐钛矿表面(1 0 1)比最稳定的金红石表面(1 1 0)更容易发生 AP 分解反应,这一点已被实验所证实。DFT 计算表明,这两种二氧化钛多晶体的活化机制是熵驱动的。
Modeling adsorption reactions of ammonium perchlorate on rutile and anatase surfaces
In this work, the effects of two TiO2 polymorphs on the decomposition of ammonium perchlorate (NH4ClO4) were studied experimentally and theoretically. The interactions between AP and various surfaces of TiO2 were modeled using density functional theory (DFT) calculations. Specifically, the adsorption of AP on three rutile surfaces (1 1 0), (1 0 0), and (0 0 1), as well as two anatase surfaces (1 0 1), and (0 0 1) were modeled using cluster models, along with the decomposition of adsorbed AP into small molecules. The optimized complexes of the AP molecule on TiO2 surfaces were very stable, indicating strong covalent and hydrogen bonding interactions, leading to highly energetic adsorption reactions. The calculated energy of adsorption (ΔEads) ranged from −120.23 to −301.98 kJ/mol, with highly exergonic calculated Gibbs free energy (ΔGads) of reaction, and highly exothermic enthalpy of reaction (ΔHads). The decomposition of adsorbed AP was also found to have very negative ΔEdec values between −199.08 and −380.73 kJ/mol. The values of ΔGdec and ΔHdec reveal exergonic and exothermic reactions. The adsorption of AP on TiO2 surfaces anticipates the heat release of decomposition, in agreement with experimental results. The most common anatase surface, (1 0 1), was predicted to be more reactive for AP decomposition than the most stable rutile surface, (1 1 0), which was confirmed by experiments. DFT calculations show the mechanism for activation of the two TiO2 polymorphs is entropy driven.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


