Junyong Gao, Mincong Wu, Jun Liao, Fanjun Meng, Changjun Chen
{"title":"在 GPU 上对一百万个分子结构进行聚类,只需几秒钟。","authors":"Junyong Gao, Mincong Wu, Jun Liao, Fanjun Meng, Changjun Chen","doi":"10.1002/jcc.27470","DOIUrl":null,"url":null,"abstract":"<p>Structure clustering is a general but time-consuming work in the study of life science. Up to now, most published tools do not support the clustering analysis on graphics processing unit (GPU) with root mean square deviation metric. In this work, we specially write codes to do the work. It supports multiple threads on multiple GPUs. To show the performance, we apply the program to a 33-residue fragment in protein Pin1 WW domain mutant. The dataset contains 1,400,000 snapshots, which are extracted from an enhanced sampling simulation and distribute widely in the conformational space. Various testing results present that our program is quite efficient. Particularly, with two NVIDIA RTX4090 GPUs and single precision data type, the clustering calculation on 1 million snapshots is completed in a few seconds (including the uploading time of data from memory to GPU and neglecting the reading time from hard disk). This is hundreds of times faster than central processing unit. Our program could be a powerful tool for fast extraction of representative states of a molecule among its thousands to millions of candidate structures.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2710-2718"},"PeriodicalIF":3.4000,"publicationDate":"2024-08-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Clustering one million molecular structures on GPU within seconds\",\"authors\":\"Junyong Gao, Mincong Wu, Jun Liao, Fanjun Meng, Changjun Chen\",\"doi\":\"10.1002/jcc.27470\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Structure clustering is a general but time-consuming work in the study of life science. Up to now, most published tools do not support the clustering analysis on graphics processing unit (GPU) with root mean square deviation metric. In this work, we specially write codes to do the work. It supports multiple threads on multiple GPUs. To show the performance, we apply the program to a 33-residue fragment in protein Pin1 WW domain mutant. The dataset contains 1,400,000 snapshots, which are extracted from an enhanced sampling simulation and distribute widely in the conformational space. Various testing results present that our program is quite efficient. Particularly, with two NVIDIA RTX4090 GPUs and single precision data type, the clustering calculation on 1 million snapshots is completed in a few seconds (including the uploading time of data from memory to GPU and neglecting the reading time from hard disk). This is hundreds of times faster than central processing unit. Our program could be a powerful tool for fast extraction of representative states of a molecule among its thousands to millions of candidate structures.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 32\",\"pages\":\"2710-2718\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-08-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27470\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27470","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Clustering one million molecular structures on GPU within seconds
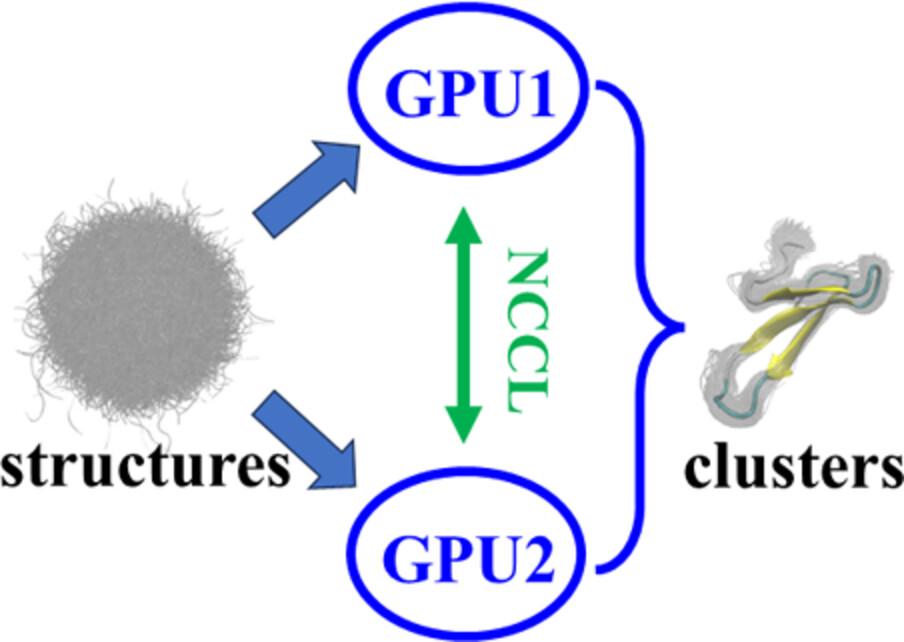
Structure clustering is a general but time-consuming work in the study of life science. Up to now, most published tools do not support the clustering analysis on graphics processing unit (GPU) with root mean square deviation metric. In this work, we specially write codes to do the work. It supports multiple threads on multiple GPUs. To show the performance, we apply the program to a 33-residue fragment in protein Pin1 WW domain mutant. The dataset contains 1,400,000 snapshots, which are extracted from an enhanced sampling simulation and distribute widely in the conformational space. Various testing results present that our program is quite efficient. Particularly, with two NVIDIA RTX4090 GPUs and single precision data type, the clustering calculation on 1 million snapshots is completed in a few seconds (including the uploading time of data from memory to GPU and neglecting the reading time from hard disk). This is hundreds of times faster than central processing unit. Our program could be a powerful tool for fast extraction of representative states of a molecule among its thousands to millions of candidate structures.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


