MGNDTI:基于多模态表征学习和门控机制的药物-靶点相互作用预测框架
IF 5.3
2区 化学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
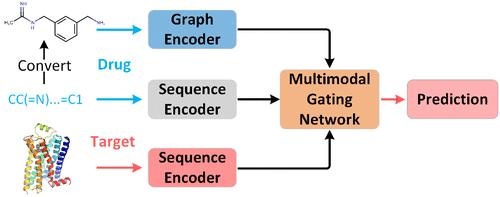
药物靶点相互作用(DTI)预测有助于加速药物发现并促进药物重新定位。现有的基于深度学习的 DTI 预测方法大多能较好地提取药物和蛋白质的鉴别特征,但很少考虑药物的多模态特征。此外,学习药物与靶点之间的相互作用表征也需要进一步探索。在此,我们基于多模态表征学习和门控机制,提出了一种用于 DTI 预测的简单 M ulti-modal G ating N etwork,即 MGNDTI。MGNDTI 首先利用不同的保持网络学习药物和靶点的序列表示。接着,它通过图卷积网络提取药物的分子图特征。随后,它设计了一个多模态门控网络,以获得药物和靶标的联合表征。最后,它建立了一个全连接网络,用于计算相互作用概率。在四种不同的实验设置下,使用四个数据集(即人类、线虫、BioSNAP 和 BindingDB)对 MGNDTI 与七种最先进的 DTI 预测模型(CPI-GNN、TransformerCPI、MolTrans、BACPI、CPGL、GIFDTI 和 FOTF-CPI)进行了基准测试。通过对 AUROC、AUPRC、准确率、F1 分数和 MCC 的评估,MGNDTI 明显优于上述七种方法。MGNDTI 是一种功能强大的 DTI 预测工具,在不同的数据集和不同的实验设置下展示了其卓越的鲁棒性和泛化能力。该工具可在 https://github.com/plhhnu/MGNDTI 免费获取。本文章由计算机程序翻译,如有差异,请以英文原文为准。
MGNDTI: A Drug-Target Interaction Prediction Framework Based on Multimodal Representation Learning and the Gating Mechanism
Drug-Target Interaction (DTI) prediction facilitates acceleration of drug discovery and promotes drug repositioning. Most existing deep learning-based DTI prediction methods can better extract discriminative features for drugs and proteins, but they rarely consider multimodal features of drugs. Moreover, learning the interaction representations between drugs and targets needs further exploration. Here, we proposed a simple M ulti-modal G ating N etwork for DTI prediction, MGNDTI, based on multimodal representation learning and the gating mechanism. MGNDTI first learns the sequence representations of drugs and targets using different retentive networks. Next, it extracts molecular graph features of drugs through a graph convolutional network. Subsequently, it devises a multimodal gating network to obtain the joint representations of drugs and targets. Finally, it builds a fully connected network for computing the interaction probability. MGNDTI was benchmarked against seven state-of-the-art DTI prediction models (CPI-GNN, TransformerCPI, MolTrans, BACPI, CPGL, GIFDTI, and FOTF-CPI) using four data sets (i.e., Human, C. elegans, BioSNAP, and BindingDB) under four different experimental settings. Through evaluation with AUROC, AUPRC, accuracy, F1 score, and MCC, MGNDTI significantly outperformed the above seven methods. MGNDTI is a powerful tool for DTI prediction, showcasing its superior robustness and generalization ability on diverse data sets and different experimental settings. It is freely available at https://github.com/plhhnu/MGNDTI.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
9.80
自引率
10.70%
发文量
529
审稿时长
1.4 months
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


