{"title":"在基于毛细管电泳的非靶向代谢组学中使用可变数据独立采集。","authors":"Saki Kiuchi, Yasuhiro Otoguro, Tomoaki Nitta, Mi Hwa Chung, Taiki Nakaya, Yuki Matsuzawa, Katsuya Ohbuchi, Kazunori Sasaki, Hiroyuki Yamamoto, Hiroshi Tsugawa","doi":"10.1021/jasms.4c00132","DOIUrl":null,"url":null,"abstract":"<p><p>Capillary electrophoresis coupled with tandem mass spectrometry (CE-MS/MS) offers advantages in peak capacity and sensitivity for metabolic profiling owing to the electroosmotic flow-based separation. However, the utilization of data-independent MS/MS acquisition (DIA) is restricted due to the absence of an optimal procedure for analytical chemistry and its related informatics framework. We assessed the mass spectral quality using two DIA techniques, namely, all-ion fragmentation (AIF) and variable DIA (vDIA), to isolate 60-800 Da precursor ions with respect to annotation rates. Our findings indicate that vDIA, coupled with the updated MS-DIAL chromatogram deconvolution algorithm, yields higher spectral matching scores and annotation rates compared to AIF. Additionally, we evaluated a linear migration time (MT) correction method using internal standards to accurately align chromatographic peaks in a data set. Postcorrection, the data set exhibited less than 0.1 min MT drifts, a difference mostly equivalent to that of conventional reverse-phase liquid chromatography techniques. Moreover, we conducted MT prediction for metabolites recorded in mass spectral libraries and metabolite structure databases containing a total of 469,870 compounds, achieving an accuracy of less than 1.5 min root mean squares. Our platform provides a peak annotation platform utilizing MT information, accurate precursor <i>m</i>/<i>z</i>, and the MS/MS spectrum recommended by the metabolomics standards initiative. Applying this procedure, we investigated metabolic alterations in lipopolysaccharide (LPS)-induced macrophages, characterizing 170 metabolites. Furthermore, we assigned metabolite information to unannotated peaks using an <i>in silico</i> structure elucidation tool, MS-FINDER. The results were integrated into the nodes in the molecular spectrum network based on the MS/MS similarity score. Consequently, we identified significantly altered metabolites in the LPS-administration group, where glycinamide ribonucleotide, not present in any spectral libraries, was newly characterized. Additionally, we retrieved metabolites of false-negative hits during the initial spectral annotation procedure. Overall, our study underscores the potential of CE-MS/MS with DIA and computational mass spectrometry techniques for metabolic profiling.</p>","PeriodicalId":672,"journal":{"name":"Journal of the American Society for Mass Spectrometry","volume":" ","pages":"2118-2127"},"PeriodicalIF":3.1000,"publicationDate":"2024-09-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Using Variable Data-Independent Acquisition for Capillary Electrophoresis-Based Untargeted Metabolomics.\",\"authors\":\"Saki Kiuchi, Yasuhiro Otoguro, Tomoaki Nitta, Mi Hwa Chung, Taiki Nakaya, Yuki Matsuzawa, Katsuya Ohbuchi, Kazunori Sasaki, Hiroyuki Yamamoto, Hiroshi Tsugawa\",\"doi\":\"10.1021/jasms.4c00132\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Capillary electrophoresis coupled with tandem mass spectrometry (CE-MS/MS) offers advantages in peak capacity and sensitivity for metabolic profiling owing to the electroosmotic flow-based separation. However, the utilization of data-independent MS/MS acquisition (DIA) is restricted due to the absence of an optimal procedure for analytical chemistry and its related informatics framework. We assessed the mass spectral quality using two DIA techniques, namely, all-ion fragmentation (AIF) and variable DIA (vDIA), to isolate 60-800 Da precursor ions with respect to annotation rates. Our findings indicate that vDIA, coupled with the updated MS-DIAL chromatogram deconvolution algorithm, yields higher spectral matching scores and annotation rates compared to AIF. Additionally, we evaluated a linear migration time (MT) correction method using internal standards to accurately align chromatographic peaks in a data set. Postcorrection, the data set exhibited less than 0.1 min MT drifts, a difference mostly equivalent to that of conventional reverse-phase liquid chromatography techniques. Moreover, we conducted MT prediction for metabolites recorded in mass spectral libraries and metabolite structure databases containing a total of 469,870 compounds, achieving an accuracy of less than 1.5 min root mean squares. Our platform provides a peak annotation platform utilizing MT information, accurate precursor <i>m</i>/<i>z</i>, and the MS/MS spectrum recommended by the metabolomics standards initiative. Applying this procedure, we investigated metabolic alterations in lipopolysaccharide (LPS)-induced macrophages, characterizing 170 metabolites. Furthermore, we assigned metabolite information to unannotated peaks using an <i>in silico</i> structure elucidation tool, MS-FINDER. The results were integrated into the nodes in the molecular spectrum network based on the MS/MS similarity score. Consequently, we identified significantly altered metabolites in the LPS-administration group, where glycinamide ribonucleotide, not present in any spectral libraries, was newly characterized. Additionally, we retrieved metabolites of false-negative hits during the initial spectral annotation procedure. Overall, our study underscores the potential of CE-MS/MS with DIA and computational mass spectrometry techniques for metabolic profiling.</p>\",\"PeriodicalId\":672,\"journal\":{\"name\":\"Journal of the American Society for Mass Spectrometry\",\"volume\":\" \",\"pages\":\"2118-2127\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-09-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the American Society for Mass Spectrometry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/jasms.4c00132\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/13 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Society for Mass Spectrometry","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/jasms.4c00132","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/13 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Using Variable Data-Independent Acquisition for Capillary Electrophoresis-Based Untargeted Metabolomics.
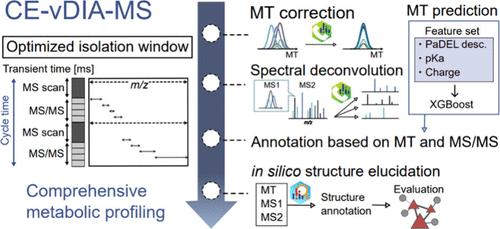
Capillary electrophoresis coupled with tandem mass spectrometry (CE-MS/MS) offers advantages in peak capacity and sensitivity for metabolic profiling owing to the electroosmotic flow-based separation. However, the utilization of data-independent MS/MS acquisition (DIA) is restricted due to the absence of an optimal procedure for analytical chemistry and its related informatics framework. We assessed the mass spectral quality using two DIA techniques, namely, all-ion fragmentation (AIF) and variable DIA (vDIA), to isolate 60-800 Da precursor ions with respect to annotation rates. Our findings indicate that vDIA, coupled with the updated MS-DIAL chromatogram deconvolution algorithm, yields higher spectral matching scores and annotation rates compared to AIF. Additionally, we evaluated a linear migration time (MT) correction method using internal standards to accurately align chromatographic peaks in a data set. Postcorrection, the data set exhibited less than 0.1 min MT drifts, a difference mostly equivalent to that of conventional reverse-phase liquid chromatography techniques. Moreover, we conducted MT prediction for metabolites recorded in mass spectral libraries and metabolite structure databases containing a total of 469,870 compounds, achieving an accuracy of less than 1.5 min root mean squares. Our platform provides a peak annotation platform utilizing MT information, accurate precursor m/z, and the MS/MS spectrum recommended by the metabolomics standards initiative. Applying this procedure, we investigated metabolic alterations in lipopolysaccharide (LPS)-induced macrophages, characterizing 170 metabolites. Furthermore, we assigned metabolite information to unannotated peaks using an in silico structure elucidation tool, MS-FINDER. The results were integrated into the nodes in the molecular spectrum network based on the MS/MS similarity score. Consequently, we identified significantly altered metabolites in the LPS-administration group, where glycinamide ribonucleotide, not present in any spectral libraries, was newly characterized. Additionally, we retrieved metabolites of false-negative hits during the initial spectral annotation procedure. Overall, our study underscores the potential of CE-MS/MS with DIA and computational mass spectrometry techniques for metabolic profiling.
期刊介绍:
The Journal of the American Society for Mass Spectrometry presents research papers covering all aspects of mass spectrometry, incorporating coverage of fields of scientific inquiry in which mass spectrometry can play a role.
Comprehensive in scope, the journal publishes papers on both fundamentals and applications of mass spectrometry. Fundamental subjects include instrumentation principles, design, and demonstration, structures and chemical properties of gas-phase ions, studies of thermodynamic properties, ion spectroscopy, chemical kinetics, mechanisms of ionization, theories of ion fragmentation, cluster ions, and potential energy surfaces. In addition to full papers, the journal offers Communications, Application Notes, and Accounts and Perspectives

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


