Buthina A. Al-Oudat, Bushra S. Abu Al fool, Suaad A. Audat, Nizar A. Al-Shar’i, Qosay A. Al-Balas, Aref Zayed, Amanda Bryant-Friedrich
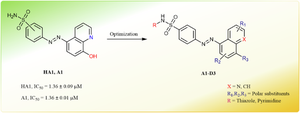
{"title":"作为抗癌候选药物的喹啉/萘基乙二醛酶-I 抑制剂的结构优化和生物学评价","authors":"Buthina A. Al-Oudat, Bushra S. Abu Al fool, Suaad A. Audat, Nizar A. Al-Shar’i, Qosay A. Al-Balas, Aref Zayed, Amanda Bryant-Friedrich","doi":"10.1007/s00044-024-03289-x","DOIUrl":null,"url":null,"abstract":"<div><p>The glyoxalase system, inherent in mammalian cells, serves as a natural detoxification mechanism that regulates cytotoxic byproducts, especially methylglyoxal (MG). Consisting of glyoxalase I (Glo-I), glyoxalase II (Glo-II), and glutathione (GSH), this system plays a vital role in managing these harmful substances. Glo-I catalyzes the rate-limiting step in MG detoxification and is found to be overexpressed in different cancer types, rendering it a promising target for novel anticancer drugs. In a previous study, a series of diazenylbenzenesulfonamide derivatives were synthesized and evaluated for their activity against Glo-I. Among these compounds, <b>HA1</b>, <b>A1</b>, and <b>HA2</b> were identified as Glo-I inhibitors with IC<sub>50</sub> values of 1.36 ± 0.09, 1.36 ± 0.01, and 1.22 ± 0.07 µM, respectively, and were subsequently chosen as lead compounds for further investigation. In the present study, the lead compounds were subjected to structural optimization to develop more potent inhibitors. Various derivatives with distinct chemical features were synthesized and tested in vitro against Glo-I to establish their structure-activity relationship and determine the key interactions within the enzyme’s active site. Several compounds exhibited potent inhibitory activity with sub-micromolar IC<sub>50</sub> values. Notably, compound (E)-8-hydroxy-5-((4-(N-(thiazol-2-yl)sulfamoyl)phenyl)diazenyl)quinoline-2-carboxylic acid (<b>B9</b>) emerged as the most potent compound, with IC<sub>50</sub> value of 0.44 ± 0.06 µM. The structure-activity relationship analysis of compound <b>B9</b> underscored the significance of the 8-hydroxyquinoline moiety as well as the sulfathiazole moiety for its inhibitory activity. To gain deeper insights into the binding modes of the compounds within the enzyme’s active site, molecular docking studies were conducted, providing enhanced and accurate predictions.</p><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":699,"journal":{"name":"Medicinal Chemistry Research","volume":"33 10","pages":"1897 - 1913"},"PeriodicalIF":2.6000,"publicationDate":"2024-07-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Structural optimization and biological evaluation of quinoline/naphthalene-based glyoxalase-I inhibitors as anti-cancer candidates\",\"authors\":\"Buthina A. Al-Oudat, Bushra S. Abu Al fool, Suaad A. Audat, Nizar A. Al-Shar’i, Qosay A. Al-Balas, Aref Zayed, Amanda Bryant-Friedrich\",\"doi\":\"10.1007/s00044-024-03289-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The glyoxalase system, inherent in mammalian cells, serves as a natural detoxification mechanism that regulates cytotoxic byproducts, especially methylglyoxal (MG). Consisting of glyoxalase I (Glo-I), glyoxalase II (Glo-II), and glutathione (GSH), this system plays a vital role in managing these harmful substances. Glo-I catalyzes the rate-limiting step in MG detoxification and is found to be overexpressed in different cancer types, rendering it a promising target for novel anticancer drugs. In a previous study, a series of diazenylbenzenesulfonamide derivatives were synthesized and evaluated for their activity against Glo-I. Among these compounds, <b>HA1</b>, <b>A1</b>, and <b>HA2</b> were identified as Glo-I inhibitors with IC<sub>50</sub> values of 1.36 ± 0.09, 1.36 ± 0.01, and 1.22 ± 0.07 µM, respectively, and were subsequently chosen as lead compounds for further investigation. In the present study, the lead compounds were subjected to structural optimization to develop more potent inhibitors. Various derivatives with distinct chemical features were synthesized and tested in vitro against Glo-I to establish their structure-activity relationship and determine the key interactions within the enzyme’s active site. Several compounds exhibited potent inhibitory activity with sub-micromolar IC<sub>50</sub> values. Notably, compound (E)-8-hydroxy-5-((4-(N-(thiazol-2-yl)sulfamoyl)phenyl)diazenyl)quinoline-2-carboxylic acid (<b>B9</b>) emerged as the most potent compound, with IC<sub>50</sub> value of 0.44 ± 0.06 µM. The structure-activity relationship analysis of compound <b>B9</b> underscored the significance of the 8-hydroxyquinoline moiety as well as the sulfathiazole moiety for its inhibitory activity. To gain deeper insights into the binding modes of the compounds within the enzyme’s active site, molecular docking studies were conducted, providing enhanced and accurate predictions.</p><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>\",\"PeriodicalId\":699,\"journal\":{\"name\":\"Medicinal Chemistry Research\",\"volume\":\"33 10\",\"pages\":\"1897 - 1913\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2024-07-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Medicinal Chemistry Research\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00044-024-03289-x\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Medicinal Chemistry Research","FirstCategoryId":"3","ListUrlMain":"https://link.springer.com/article/10.1007/s00044-024-03289-x","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Structural optimization and biological evaluation of quinoline/naphthalene-based glyoxalase-I inhibitors as anti-cancer candidates
The glyoxalase system, inherent in mammalian cells, serves as a natural detoxification mechanism that regulates cytotoxic byproducts, especially methylglyoxal (MG). Consisting of glyoxalase I (Glo-I), glyoxalase II (Glo-II), and glutathione (GSH), this system plays a vital role in managing these harmful substances. Glo-I catalyzes the rate-limiting step in MG detoxification and is found to be overexpressed in different cancer types, rendering it a promising target for novel anticancer drugs. In a previous study, a series of diazenylbenzenesulfonamide derivatives were synthesized and evaluated for their activity against Glo-I. Among these compounds, HA1, A1, and HA2 were identified as Glo-I inhibitors with IC50 values of 1.36 ± 0.09, 1.36 ± 0.01, and 1.22 ± 0.07 µM, respectively, and were subsequently chosen as lead compounds for further investigation. In the present study, the lead compounds were subjected to structural optimization to develop more potent inhibitors. Various derivatives with distinct chemical features were synthesized and tested in vitro against Glo-I to establish their structure-activity relationship and determine the key interactions within the enzyme’s active site. Several compounds exhibited potent inhibitory activity with sub-micromolar IC50 values. Notably, compound (E)-8-hydroxy-5-((4-(N-(thiazol-2-yl)sulfamoyl)phenyl)diazenyl)quinoline-2-carboxylic acid (B9) emerged as the most potent compound, with IC50 value of 0.44 ± 0.06 µM. The structure-activity relationship analysis of compound B9 underscored the significance of the 8-hydroxyquinoline moiety as well as the sulfathiazole moiety for its inhibitory activity. To gain deeper insights into the binding modes of the compounds within the enzyme’s active site, molecular docking studies were conducted, providing enhanced and accurate predictions.
期刊介绍:
Medicinal Chemistry Research (MCRE) publishes papers on a wide range of topics, favoring research with significant, new, and up-to-date information. Although the journal has a demanding peer review process, MCRE still boasts rapid publication, due in part, to the length of the submissions. The journal publishes significant research on various topics, many of which emphasize the structure-activity relationships of molecular biology.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


