Qingqi Lin, Yair Dorsett, Ali Mirza, Helen Tremlett, Laura Piccio, Erin E Longbrake, Siobhan Ni Choileain, David A Hafler, Laura M Cox, Howard L Weiner, Takashi Yamamura, Kun Chen, Yufeng Wu, Yanjiao Zhou
{"title":"元分析确定了与多发性硬化症有关的常见肠道微生物群。","authors":"Qingqi Lin, Yair Dorsett, Ali Mirza, Helen Tremlett, Laura Piccio, Erin E Longbrake, Siobhan Ni Choileain, David A Hafler, Laura M Cox, Howard L Weiner, Takashi Yamamura, Kun Chen, Yufeng Wu, Yanjiao Zhou","doi":"10.1186/s13073-024-01364-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Previous studies have identified a diverse group of microbial taxa that differ between patients with multiple sclerosis (MS) and the healthy population. However, interpreting findings on MS-associated microbiota is challenging, as there is no true consensus. It is unclear whether there is gut microbiota commonly altered in MS across studies.</p><p><strong>Methods: </strong>To answer this, we performed a meta-analysis based on the 16S rRNA gene sequencing data from seven geographically and technically diverse studies comprising a total of 524 adult subjects (257 MS and 267 healthy controls). Analysis was conducted for each individual study after reprocessing the data and also by combining all data together. The blocked Wilcoxon rank-sum test and linear mixed-effects regression were used to identify differences in microbial composition and diversity between MS and healthy controls. Network analysis was conducted to identify bacterial correlations. A leave-one-out sensitivity analysis was performed to ensure the robustness of the findings.</p><p><strong>Results: </strong>The microbiome community structure was significantly different between studies. Re-analysis of data from individual studies revealed a lower relative abundance of Prevotella in MS across studies, compared to controls. Meta-analysis found that although alpha and beta diversity did not differ between MS and controls, a higher abundance of Actinomyces and a lower abundance of Faecalibacterium were reproducibly associated with MS. Additionally, network analysis revealed that the recognized negative Bacteroides-Prevotella correlation in controls was disrupted in patients with MS.</p><p><strong>Conclusions: </strong>Our meta-analysis identified common gut microbiota associated with MS across geographically and technically diverse studies.</p>","PeriodicalId":12645,"journal":{"name":"Genome Medicine","volume":"16 1","pages":"94"},"PeriodicalIF":10.4000,"publicationDate":"2024-07-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11293023/pdf/","citationCount":"0","resultStr":"{\"title\":\"Meta-analysis identifies common gut microbiota associated with multiple sclerosis.\",\"authors\":\"Qingqi Lin, Yair Dorsett, Ali Mirza, Helen Tremlett, Laura Piccio, Erin E Longbrake, Siobhan Ni Choileain, David A Hafler, Laura M Cox, Howard L Weiner, Takashi Yamamura, Kun Chen, Yufeng Wu, Yanjiao Zhou\",\"doi\":\"10.1186/s13073-024-01364-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Previous studies have identified a diverse group of microbial taxa that differ between patients with multiple sclerosis (MS) and the healthy population. However, interpreting findings on MS-associated microbiota is challenging, as there is no true consensus. It is unclear whether there is gut microbiota commonly altered in MS across studies.</p><p><strong>Methods: </strong>To answer this, we performed a meta-analysis based on the 16S rRNA gene sequencing data from seven geographically and technically diverse studies comprising a total of 524 adult subjects (257 MS and 267 healthy controls). Analysis was conducted for each individual study after reprocessing the data and also by combining all data together. The blocked Wilcoxon rank-sum test and linear mixed-effects regression were used to identify differences in microbial composition and diversity between MS and healthy controls. Network analysis was conducted to identify bacterial correlations. A leave-one-out sensitivity analysis was performed to ensure the robustness of the findings.</p><p><strong>Results: </strong>The microbiome community structure was significantly different between studies. Re-analysis of data from individual studies revealed a lower relative abundance of Prevotella in MS across studies, compared to controls. Meta-analysis found that although alpha and beta diversity did not differ between MS and controls, a higher abundance of Actinomyces and a lower abundance of Faecalibacterium were reproducibly associated with MS. Additionally, network analysis revealed that the recognized negative Bacteroides-Prevotella correlation in controls was disrupted in patients with MS.</p><p><strong>Conclusions: </strong>Our meta-analysis identified common gut microbiota associated with MS across geographically and technically diverse studies.</p>\",\"PeriodicalId\":12645,\"journal\":{\"name\":\"Genome Medicine\",\"volume\":\"16 1\",\"pages\":\"94\"},\"PeriodicalIF\":10.4000,\"publicationDate\":\"2024-07-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11293023/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genome Medicine\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13073-024-01364-x\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genome Medicine","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13073-024-01364-x","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Meta-analysis identifies common gut microbiota associated with multiple sclerosis.
Background: Previous studies have identified a diverse group of microbial taxa that differ between patients with multiple sclerosis (MS) and the healthy population. However, interpreting findings on MS-associated microbiota is challenging, as there is no true consensus. It is unclear whether there is gut microbiota commonly altered in MS across studies.
Methods: To answer this, we performed a meta-analysis based on the 16S rRNA gene sequencing data from seven geographically and technically diverse studies comprising a total of 524 adult subjects (257 MS and 267 healthy controls). Analysis was conducted for each individual study after reprocessing the data and also by combining all data together. The blocked Wilcoxon rank-sum test and linear mixed-effects regression were used to identify differences in microbial composition and diversity between MS and healthy controls. Network analysis was conducted to identify bacterial correlations. A leave-one-out sensitivity analysis was performed to ensure the robustness of the findings.
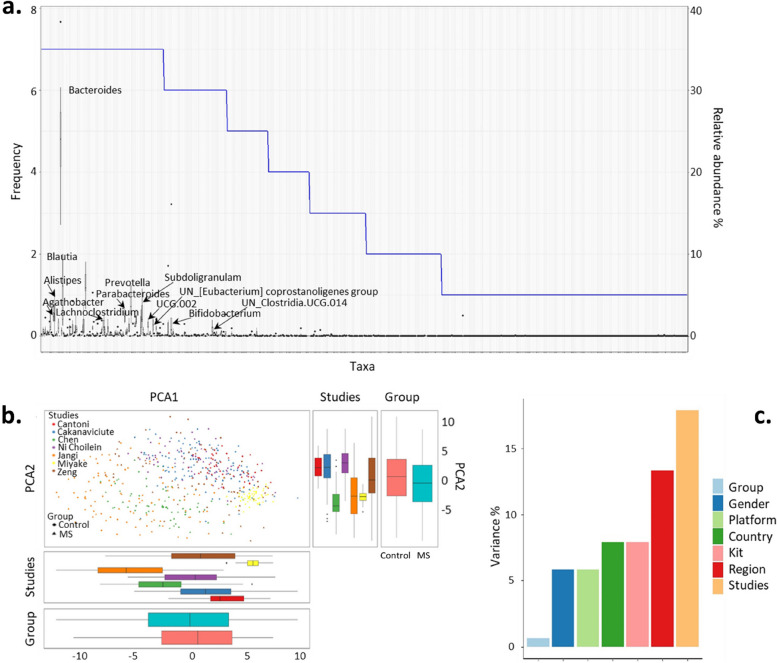
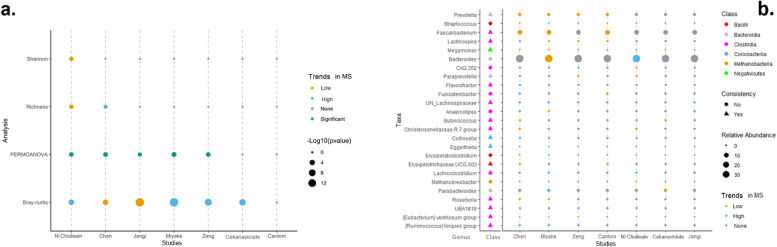
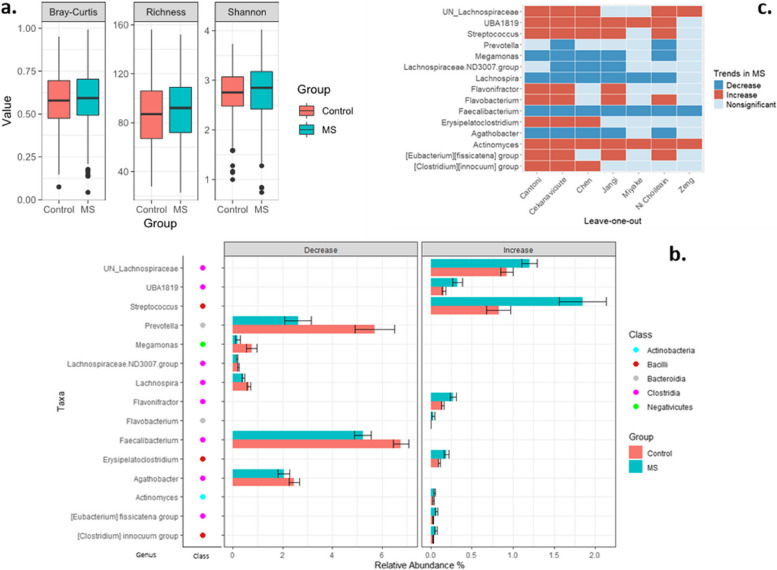
Results: The microbiome community structure was significantly different between studies. Re-analysis of data from individual studies revealed a lower relative abundance of Prevotella in MS across studies, compared to controls. Meta-analysis found that although alpha and beta diversity did not differ between MS and controls, a higher abundance of Actinomyces and a lower abundance of Faecalibacterium were reproducibly associated with MS. Additionally, network analysis revealed that the recognized negative Bacteroides-Prevotella correlation in controls was disrupted in patients with MS.
Conclusions: Our meta-analysis identified common gut microbiota associated with MS across geographically and technically diverse studies.
期刊介绍:
Genome Medicine is an open access journal that publishes outstanding research applying genetics, genomics, and multi-omics to understand, diagnose, and treat disease. Bridging basic science and clinical research, it covers areas such as cancer genomics, immuno-oncology, immunogenomics, infectious disease, microbiome, neurogenomics, systems medicine, clinical genomics, gene therapies, precision medicine, and clinical trials. The journal publishes original research, methods, software, and reviews to serve authors and promote broad interest and importance in the field.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


