{"title":"关于乙腈中阴极条件下酰肼异解键裂解的 DFT 研究。","authors":"Mark A. W. Lawrence","doi":"10.1007/s00894-024-06082-0","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Hydrazones have been studied for a myriad of chemical and physiochemical properties, such as sensors, chelators and numerous biological activities. Experimental data indicates that hydrazones are unstable under cathodic potentials irrespective of the solvent. The single electron reduction of hydrazones to produce radical anions result in unstable species that cleaves at the N–N bond in a heterolytic manner. The literature has proposed a mechanism favouring the radical on the imine moiety, however in this study DFT calculations suggest the radical on the amine product is more likely upon bond cleavage. This has implications on electrochemical mechanisms, and the active molecule in biological studies viz the method of delivery to target areas.</p><h3>Methods</h3><p>Density functional theory calculations were carried out using the GAMESS software package. The structures were optimized in the gas phase (B3LYP/6-31G(d,p)) as indicated by the absence of imaginary frequencies in the Hessian, and in CH<sub>3</sub>CN (B3LYP/6-31G(d,p)/SMD) with the Pople polarization functions. As a comparison, selected pathways were fully optimized using PBE0/6-31G(d,p) and PBE0/6-31G(d,p)/SMD for gas phase and CH<sub>3</sub>CN, respectively with the Pople polarization functions. The values were not significantly different (< 5% difference). As such only the B3LYP is evaluation is discussed.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 8","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2024-07-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A DFT study on the heterolytic bond cleavage of hydrazones under cathodic conditions in acetonitrile\",\"authors\":\"Mark A. W. Lawrence\",\"doi\":\"10.1007/s00894-024-06082-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>Hydrazones have been studied for a myriad of chemical and physiochemical properties, such as sensors, chelators and numerous biological activities. Experimental data indicates that hydrazones are unstable under cathodic potentials irrespective of the solvent. The single electron reduction of hydrazones to produce radical anions result in unstable species that cleaves at the N–N bond in a heterolytic manner. The literature has proposed a mechanism favouring the radical on the imine moiety, however in this study DFT calculations suggest the radical on the amine product is more likely upon bond cleavage. This has implications on electrochemical mechanisms, and the active molecule in biological studies viz the method of delivery to target areas.</p><h3>Methods</h3><p>Density functional theory calculations were carried out using the GAMESS software package. The structures were optimized in the gas phase (B3LYP/6-31G(d,p)) as indicated by the absence of imaginary frequencies in the Hessian, and in CH<sub>3</sub>CN (B3LYP/6-31G(d,p)/SMD) with the Pople polarization functions. As a comparison, selected pathways were fully optimized using PBE0/6-31G(d,p) and PBE0/6-31G(d,p)/SMD for gas phase and CH<sub>3</sub>CN, respectively with the Pople polarization functions. The values were not significantly different (< 5% difference). As such only the B3LYP is evaluation is discussed.</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"30 8\",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-07-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-024-06082-0\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06082-0","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
A DFT study on the heterolytic bond cleavage of hydrazones under cathodic conditions in acetonitrile
Context
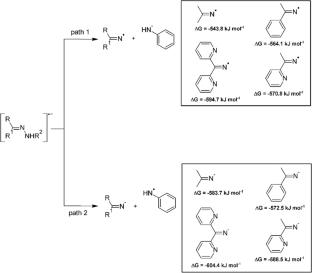
Hydrazones have been studied for a myriad of chemical and physiochemical properties, such as sensors, chelators and numerous biological activities. Experimental data indicates that hydrazones are unstable under cathodic potentials irrespective of the solvent. The single electron reduction of hydrazones to produce radical anions result in unstable species that cleaves at the N–N bond in a heterolytic manner. The literature has proposed a mechanism favouring the radical on the imine moiety, however in this study DFT calculations suggest the radical on the amine product is more likely upon bond cleavage. This has implications on electrochemical mechanisms, and the active molecule in biological studies viz the method of delivery to target areas.
Methods
Density functional theory calculations were carried out using the GAMESS software package. The structures were optimized in the gas phase (B3LYP/6-31G(d,p)) as indicated by the absence of imaginary frequencies in the Hessian, and in CH3CN (B3LYP/6-31G(d,p)/SMD) with the Pople polarization functions. As a comparison, selected pathways were fully optimized using PBE0/6-31G(d,p) and PBE0/6-31G(d,p)/SMD for gas phase and CH3CN, respectively with the Pople polarization functions. The values were not significantly different (< 5% difference). As such only the B3LYP is evaluation is discussed.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


