{"title":"模拟黄素光谱学和光物理学的电子结构方法:多参量、TD-DFT 和单参量波函数方法的比较。","authors":"Mohammad Pabel Kabir, Paulami Ghosh, Samer Gozem","doi":"10.1021/acs.jpcb.4c03748","DOIUrl":null,"url":null,"abstract":"<p><p>The use of flavins and flavoproteins in photocatalytic, sensing, and biotechnological applications has led to a growing interest in computationally modeling the excited-state electronic structure and photophysics of flavin. However, there is limited consensus regarding which computational methods are appropriate for modeling flavin's photophysics. We compare the energies of low-lying excited states of flavin computed with time-dependent density functional theory (TD-DFT), equation-of-motion coupled cluster (EOM-EE-CCSD), scaled opposite-spin configuration interaction [SOS-CIS(D)], multiconfiguration pair-density functional theory (MC-PDFT), and several multireference perturbation theory (MR-PT2) methods. In the first part, we focus on excitation energies of the first singlet excited state (S<sub>1</sub>) of five different redox and protonation states of flavin, with the goal of finding a suitable active space for MR-PT2 calculations. In the second part, we construct two sets of one-dimensional potential energy surfaces connecting the S<sub>0</sub> and S<sub>1</sub> equilibrium geometries (S<sub>0</sub>-S<sub>1</sub> path) and the S<sub>1</sub> (π,π*) and S<sub>2</sub> (<i>n</i>,π*) equilibrium geometries (S<sub>1</sub>-S<sub>2</sub> path). The first path therefore follows a Franck-Condon active mode of flavin while the second path maps crossings points between low-lying singlet and triplet states in flavin. We discuss the similarities and differences in the TD-DFT, EOM-EE-CCSD, SOS-CIS(D), MC-PDFT and MR-PT2 energy profiles along these paths. We find that (TD-)DFT methods are suitable for applications such as simulating the spectra of flavins but are inconsistent with several other methods when used for some geometry optimizations and when describing the energetics of dark (<i>n</i>,π*) states. MR-PT2 methods show promise for the simulation of flavin's low-lying excited states, but the selection of orbitals for the active space and the number of roots used for state averaging must be done carefully to avoid artifacts. Some properties, such as the intersystem crossing geometry and energy between the S<sub>1</sub> (π,π*) and T<sub>2</sub> (<i>n</i>,π*) states, may require additional benchmarking before they can be determined quantitatively.</p>","PeriodicalId":60,"journal":{"name":"The Journal of Physical Chemistry B","volume":" ","pages":"7545-7557"},"PeriodicalIF":2.9000,"publicationDate":"2024-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11317985/pdf/","citationCount":"0","resultStr":"{\"title\":\"Electronic Structure Methods for Simulating Flavin's Spectroscopy and Photophysics: Comparison of Multi-reference, TD-DFT, and Single-Reference Wave Function Methods.\",\"authors\":\"Mohammad Pabel Kabir, Paulami Ghosh, Samer Gozem\",\"doi\":\"10.1021/acs.jpcb.4c03748\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The use of flavins and flavoproteins in photocatalytic, sensing, and biotechnological applications has led to a growing interest in computationally modeling the excited-state electronic structure and photophysics of flavin. However, there is limited consensus regarding which computational methods are appropriate for modeling flavin's photophysics. We compare the energies of low-lying excited states of flavin computed with time-dependent density functional theory (TD-DFT), equation-of-motion coupled cluster (EOM-EE-CCSD), scaled opposite-spin configuration interaction [SOS-CIS(D)], multiconfiguration pair-density functional theory (MC-PDFT), and several multireference perturbation theory (MR-PT2) methods. In the first part, we focus on excitation energies of the first singlet excited state (S<sub>1</sub>) of five different redox and protonation states of flavin, with the goal of finding a suitable active space for MR-PT2 calculations. In the second part, we construct two sets of one-dimensional potential energy surfaces connecting the S<sub>0</sub> and S<sub>1</sub> equilibrium geometries (S<sub>0</sub>-S<sub>1</sub> path) and the S<sub>1</sub> (π,π*) and S<sub>2</sub> (<i>n</i>,π*) equilibrium geometries (S<sub>1</sub>-S<sub>2</sub> path). The first path therefore follows a Franck-Condon active mode of flavin while the second path maps crossings points between low-lying singlet and triplet states in flavin. We discuss the similarities and differences in the TD-DFT, EOM-EE-CCSD, SOS-CIS(D), MC-PDFT and MR-PT2 energy profiles along these paths. We find that (TD-)DFT methods are suitable for applications such as simulating the spectra of flavins but are inconsistent with several other methods when used for some geometry optimizations and when describing the energetics of dark (<i>n</i>,π*) states. MR-PT2 methods show promise for the simulation of flavin's low-lying excited states, but the selection of orbitals for the active space and the number of roots used for state averaging must be done carefully to avoid artifacts. Some properties, such as the intersystem crossing geometry and energy between the S<sub>1</sub> (π,π*) and T<sub>2</sub> (<i>n</i>,π*) states, may require additional benchmarking before they can be determined quantitatively.</p>\",\"PeriodicalId\":60,\"journal\":{\"name\":\"The Journal of Physical Chemistry B\",\"volume\":\" \",\"pages\":\"7545-7557\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-08-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11317985/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry B\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpcb.4c03748\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/7/29 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcb.4c03748","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/29 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Electronic Structure Methods for Simulating Flavin's Spectroscopy and Photophysics: Comparison of Multi-reference, TD-DFT, and Single-Reference Wave Function Methods.
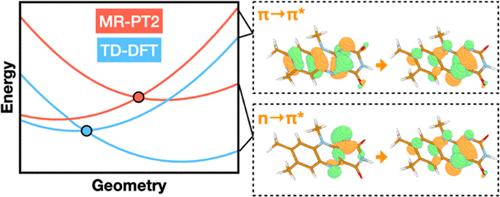
The use of flavins and flavoproteins in photocatalytic, sensing, and biotechnological applications has led to a growing interest in computationally modeling the excited-state electronic structure and photophysics of flavin. However, there is limited consensus regarding which computational methods are appropriate for modeling flavin's photophysics. We compare the energies of low-lying excited states of flavin computed with time-dependent density functional theory (TD-DFT), equation-of-motion coupled cluster (EOM-EE-CCSD), scaled opposite-spin configuration interaction [SOS-CIS(D)], multiconfiguration pair-density functional theory (MC-PDFT), and several multireference perturbation theory (MR-PT2) methods. In the first part, we focus on excitation energies of the first singlet excited state (S1) of five different redox and protonation states of flavin, with the goal of finding a suitable active space for MR-PT2 calculations. In the second part, we construct two sets of one-dimensional potential energy surfaces connecting the S0 and S1 equilibrium geometries (S0-S1 path) and the S1 (π,π*) and S2 (n,π*) equilibrium geometries (S1-S2 path). The first path therefore follows a Franck-Condon active mode of flavin while the second path maps crossings points between low-lying singlet and triplet states in flavin. We discuss the similarities and differences in the TD-DFT, EOM-EE-CCSD, SOS-CIS(D), MC-PDFT and MR-PT2 energy profiles along these paths. We find that (TD-)DFT methods are suitable for applications such as simulating the spectra of flavins but are inconsistent with several other methods when used for some geometry optimizations and when describing the energetics of dark (n,π*) states. MR-PT2 methods show promise for the simulation of flavin's low-lying excited states, but the selection of orbitals for the active space and the number of roots used for state averaging must be done carefully to avoid artifacts. Some properties, such as the intersystem crossing geometry and energy between the S1 (π,π*) and T2 (n,π*) states, may require additional benchmarking before they can be determined quantitatively.
期刊介绍:
An essential criterion for acceptance of research articles in the journal is that they provide new physical insight. Please refer to the New Physical Insights virtual issue on what constitutes new physical insight. Manuscripts that are essentially reporting data or applications of data are, in general, not suitable for publication in JPC B.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


