{"title":"具有苯基苯并噻唑取代基的发光磷胍的合成与构象特征","authors":"M. Y. Afonin, S. N. Konchenko, T. S. Sukhikh","doi":"10.1134/S0022476624070138","DOIUrl":null,"url":null,"abstract":"<p>We report a three-stage scheme (1) Pbt–NH<sub>2</sub> + CS<sub>2</sub> → (Pbt–NH)<sub>2</sub>C=S (<b>1</b>) (Pbt = 4-(1′,3′-benzothiazole-2′-yl)phenyl), (2) <b>1</b> + PPh<sub>3</sub> + I<sub>2</sub> → Pbt–N=C=N–Pbt (<b>2</b>), (3) <b>2</b> + Ph<sub>2</sub>PH → (Pbt–N)(Pbt–NH)CPPh<sub>2</sub> (<b>3</b>) for the synthesis of a novel luminescent phosphoguanidine <b>3</b> with an unprecedentedly high yield (90%) at the last catalyst-free stage. It is demonstrated by the density functional theory (DFT) method that high reactivity of <b>2</b>, leading to such an yield, is explained by a high electrostatic potential at the central carbon atom. For <b>3,</b> two polymorphs <b>3α</b>, <b>3β</b> and a solvatomorph <b>3γ</b>·THF are prepared. The structures of <b>2</b>, <b>3α</b>, <b>3β</b>, and <b>3γ</b>·THF are determined by single-crystal XRD. The tendency of crystals of different phenylbenzothiazole derivatives to form different conformations is explained by the computational (DFT) data indicating that the energy change of the molecule of <b>3</b> considered as a function of the torsion angle between phenyl and benzothiazole fragments does not exceed 2 kJ/mol in the –15…30° range. Photophysical properties of <b>3β</b> and <b>3γ</b>·THF phases are studied. It is shown that these compounds exhibit photoluminescence with an emission maximum at 510 nm.</p>","PeriodicalId":668,"journal":{"name":"Journal of Structural Chemistry","volume":"65 7","pages":"1420 - 1431"},"PeriodicalIF":1.2000,"publicationDate":"2024-07-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Synthesis and Conformational Features of Luminescent Phosphoguanidine with a Phenylbenzothiazole Substituent\",\"authors\":\"M. Y. Afonin, S. N. Konchenko, T. S. Sukhikh\",\"doi\":\"10.1134/S0022476624070138\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>We report a three-stage scheme (1) Pbt–NH<sub>2</sub> + CS<sub>2</sub> → (Pbt–NH)<sub>2</sub>C=S (<b>1</b>) (Pbt = 4-(1′,3′-benzothiazole-2′-yl)phenyl), (2) <b>1</b> + PPh<sub>3</sub> + I<sub>2</sub> → Pbt–N=C=N–Pbt (<b>2</b>), (3) <b>2</b> + Ph<sub>2</sub>PH → (Pbt–N)(Pbt–NH)CPPh<sub>2</sub> (<b>3</b>) for the synthesis of a novel luminescent phosphoguanidine <b>3</b> with an unprecedentedly high yield (90%) at the last catalyst-free stage. It is demonstrated by the density functional theory (DFT) method that high reactivity of <b>2</b>, leading to such an yield, is explained by a high electrostatic potential at the central carbon atom. For <b>3,</b> two polymorphs <b>3α</b>, <b>3β</b> and a solvatomorph <b>3γ</b>·THF are prepared. The structures of <b>2</b>, <b>3α</b>, <b>3β</b>, and <b>3γ</b>·THF are determined by single-crystal XRD. The tendency of crystals of different phenylbenzothiazole derivatives to form different conformations is explained by the computational (DFT) data indicating that the energy change of the molecule of <b>3</b> considered as a function of the torsion angle between phenyl and benzothiazole fragments does not exceed 2 kJ/mol in the –15…30° range. Photophysical properties of <b>3β</b> and <b>3γ</b>·THF phases are studied. It is shown that these compounds exhibit photoluminescence with an emission maximum at 510 nm.</p>\",\"PeriodicalId\":668,\"journal\":{\"name\":\"Journal of Structural Chemistry\",\"volume\":\"65 7\",\"pages\":\"1420 - 1431\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2024-07-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Structural Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1134/S0022476624070138\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Structural Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1134/S0022476624070138","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
Synthesis and Conformational Features of Luminescent Phosphoguanidine with a Phenylbenzothiazole Substituent
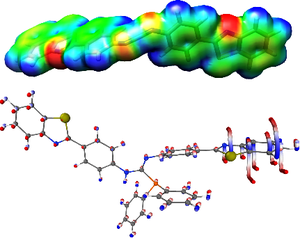
We report a three-stage scheme (1) Pbt–NH2 + CS2 → (Pbt–NH)2C=S (1) (Pbt = 4-(1′,3′-benzothiazole-2′-yl)phenyl), (2) 1 + PPh3 + I2 → Pbt–N=C=N–Pbt (2), (3) 2 + Ph2PH → (Pbt–N)(Pbt–NH)CPPh2 (3) for the synthesis of a novel luminescent phosphoguanidine 3 with an unprecedentedly high yield (90%) at the last catalyst-free stage. It is demonstrated by the density functional theory (DFT) method that high reactivity of 2, leading to such an yield, is explained by a high electrostatic potential at the central carbon atom. For 3, two polymorphs 3α, 3β and a solvatomorph 3γ·THF are prepared. The structures of 2, 3α, 3β, and 3γ·THF are determined by single-crystal XRD. The tendency of crystals of different phenylbenzothiazole derivatives to form different conformations is explained by the computational (DFT) data indicating that the energy change of the molecule of 3 considered as a function of the torsion angle between phenyl and benzothiazole fragments does not exceed 2 kJ/mol in the –15…30° range. Photophysical properties of 3β and 3γ·THF phases are studied. It is shown that these compounds exhibit photoluminescence with an emission maximum at 510 nm.
期刊介绍:
Journal is an interdisciplinary publication covering all aspects of structural chemistry, including the theory of molecular structure and chemical bond; the use of physical methods to study the electronic and spatial structure of chemical species; structural features of liquids, solutions, surfaces, supramolecular systems, nano- and solid materials; and the crystal structure of solids.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


