基于表面活性剂的药物制剂的口服吸收:分子溶解药物对生物利用率的影响。
IF 3.7
3区 医学
Q2 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
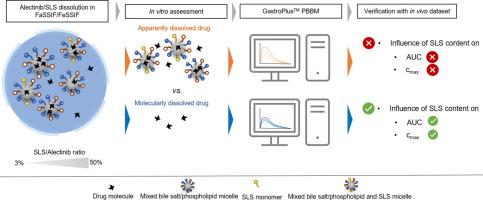
为确保口服溶解性差的药物被充分吸收,通常需要使用药物制剂。虽然这些制剂通常会增加药物的表观溶解度,但人们普遍认为,只有分子溶解的药物(即药物的游离部分)才容易被直接吸收,而胶体相关药物的渗透程度并不相同。在本研究中,我们旨在比较分子溶解的药物浓度和表面溶解的药物浓度(即分子药物和胶体药物的总和)对水溶性较差的药物化合物阿来替尼口服吸收的影响。我们制备了阿来替尼(Alectinib)与相对于剂量分别为50%、25%、12.5%和3%的十二烷基硫酸钠(SLS)的混合物,并在模拟进食和空腹状态下进行了小规模溶出试验。同时分别使用微透析和离心/过滤取样方法评估了药物的分子浓度和表观溶解浓度。这些数据是体外-体内相关性(IVIVC)的基础,也是 GastroPlusTM 生理生物制药模型(PBBM)的输入数据。结果表明,随着 SLS 含量的增加,FeSSIF 和 FaSSIF 中的表观溶解药物呈线性增加,因此,根据表观溶解药物预测,50% SLS 制剂的体内性能将优于所有其他制剂。然而,与一般预期不同的是,游离(分子溶解)药物浓度也随 SLS 浓度的变化而变化,但变化程度较小。通过系统比较配方中不同 SLS 含量和餐前状态下的溶解和游离药物溶解模式,可以深入了解配方的复杂溶解/过饱和、胶束化和沉淀行为。在将体外数据集与生物等效性研究中的人体药代动力学数据进行比较时,结果表明使用分子溶解药物可提高 IVIVC。将体外溶解数据集纳入 GastroPlusTM PBBM 后,表面溶解的药物浓度导致对血浆浓度的预测明显偏高,同时也错误地预测了 SLS 对全身暴露的影响。相反,将分子溶解的药物(即游离部分)作为模型输入,预测的血浆浓度-时间曲线与所有制剂在进食和空腹条件下的观察数据非常吻合。通过将先进的体外评估与 PBBM 结合起来,本研究证实了只有分子溶解的药物而非胶体相关药物可被直接吸收。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Oral Absorption from Surfactant-Based Drug Formulations: The Impact of Molecularly Dissolved Drug on Bioavailability
Enabling drug formulations are often required to ensure sufficient absorption after oral administration of poorly soluble drugs. While these formulations typically increase the apparent solubility of the drug, it is widely acknowledged that only molecularly dissolved, i.e., free fraction of the drug, is prone for direct absorption, while colloid-associated drug does not permeate to the same extent.
In the present study, we aimed at comparing the effect of molecularly and apparently (i.e., the sum of molecularly and colloid-associated drug) dissolved drug concentrations on the oral absorption of a poorly water-soluble drug compound, Alectinib. Mixtures of Alectinib and respectively 50 %, 25 %, 12.5 %, and 3 % sodium lauryl sulfate (SLS) relative to the dose were prepared and small-scale dissolution tests were performed under simulated fed and fasted state conditions. Both the molecularly and apparently dissolved drug concentrations were assessed in parallel using microdialysis and centrifugation/filtration sampling, respectively. The data served as the basis for an in vitro-in vivo correlation (IVIVC) and as input for a GastroPlusTM physiologically-based biopharmaceutics model (PBBM).
It was shown that with increasing the content of SLS the apparently dissolved drug in FeSSIF and FaSSIF increased to a linear extent and thus, the predicted in vivo performance of the 50 % SLS formulation, based on apparently dissolved drug, would outperform all other formulations. Against common expectation, however, the free (molecularly dissolved) drug concentrations were found to vary with SLS concentrations as well, yet to a minor extent. A systematic comparison of solubilized and free drug dissolution patterns at different SLS contents of the formulations and prandial states allowed for interesting insights into the complex dissolution-/supersaturation-, micellization-, and precipitation-behavior of the formulations. When comparing the in vitro datasets with human pharmacokinetic data from a bioequivalence study, it was shown that the use of molecularly dissolved drug resulted in an improved IVIVC.
By incorporating the in vitro dissolution datasets into the GastroPlusTM PBBM, the apparently dissolved drug concentrations resulted in both, a remarkable overprediction of plasma concentrations as well as a misprediction of the influence of SLS on systemic exposure. In contrast, by using the molecularly dissolved drug (i.e., free fraction) as the model input, the predicted plasma concentration-time profiles were in excellent agreement with observed data for all formulations under both fed and fasted conditions.
By combining an advanced in vitro assessment with PBBM, the present study confirmed that only the molecularly dissolved drug, and not the colloid-associated drug, is available for direct absorption.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
7.30
自引率
13.20%
发文量
367
审稿时长
33 days
期刊介绍:
The Journal of Pharmaceutical Sciences will publish original research papers, original research notes, invited topical reviews (including Minireviews), and editorial commentary and news. The area of focus shall be concepts in basic pharmaceutical science and such topics as chemical processing of pharmaceuticals, including crystallization, lyophilization, chemical stability of drugs, pharmacokinetics, biopharmaceutics, pharmacodynamics, pro-drug developments, metabolic disposition of bioactive agents, dosage form design, protein-peptide chemistry and biotechnology specifically as these relate to pharmaceutical technology, and targeted drug delivery.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


