Christos Varelas, Efthymia Vlachaki, Philippos Klonizakis, Despoina Pantelidou, Fani Minti, Michael Diamantidis, Nikolaos Sabanis, Evdoxia Koravou, Ioanna Christodoulou, Despina Papadopoulou, Stamatia Theodoridou, Tasoula Touloumenidou, Apostolia Papalexandri, Ioanna Sakellari, Sofia Vakalopoulou, Vasilis Perifanis, George Vassilopoulos, Ioannis Mitroulis, Eleni Gavriilaki
{"title":"镰状细胞病及其并发症中补体激活和血栓性炎症的前瞻性研究。","authors":"Christos Varelas, Efthymia Vlachaki, Philippos Klonizakis, Despoina Pantelidou, Fani Minti, Michael Diamantidis, Nikolaos Sabanis, Evdoxia Koravou, Ioanna Christodoulou, Despina Papadopoulou, Stamatia Theodoridou, Tasoula Touloumenidou, Apostolia Papalexandri, Ioanna Sakellari, Sofia Vakalopoulou, Vasilis Perifanis, George Vassilopoulos, Ioannis Mitroulis, Eleni Gavriilaki","doi":"10.1002/hem3.135","DOIUrl":null,"url":null,"abstract":"<p>Sickle cell disease (SCD) results from mutations in the β-globin gene, producing abnormal hemoglobin S (HbS) and leading to complications causing significant morbidity and mortality.<span><sup>1</sup></span> One of the hallmark consequences of SCD is the occurrence of vaso-occlusive crises (VOCs), which arise from the interplay of factors in the disease's pathophysiology, involving abnormal hemoglobin polymerization, inflammation, endothelial dysfunction, and activation of the immune system, culminating in the painful obstruction of blood vessels by sickled red blood cells, that tend to obstruct blood vessels, leading to reduced blood flow and oxygen supply. This vicious cycle of ischemia followed by reperfusion constitutes the ischemia-reperfusion model.<span><sup>2</sup></span></p><p>The complement system, a complex defense mechanism, is implicated in various diseases through unregulated activation.<span><sup>3</sup></span> However, diagnostic challenges hinder patient selection for complement inhibition.<span><sup>4</sup></span> Preliminary data from our group using novel assays indicate complement activation even at a steady state in a limited patient population.<span><sup>5</sup></span></p><p>Limited information exists on additional markers in the complement activation and endothelial dysfunction cycle in SCD. Neutrophil extracellular traps (NETs), indicative of thromboinflammation, are elevated in SCD patients, even during steady state.<span><sup>6</sup></span> ADAMTS13 (A Disintegrin and Metalloproteinase with Thrombospondin motifs), studied for its role in SCD vasculopathy, shows conflicting results as a potential biomarker.<span><sup>7, 8</sup></span> Genetic variants and autoantibodies leading to unregulated complement activation are implicated in the pathogenesis of various human diseases.<span><sup>9</sup></span></p><p>Despite the lack of specific biomarkers or targeted treatments for crises, our hypothesis posits the presence of complement activation and thromboinflammation in SCD, particularly during complications, with distinct yet unexplored clinical or genetic features in these patients.</p><p>Our study's methods regarding patient population, observation period, functional assays, and genetic, bioinformatic, and statistical analysis are demonstrated in supplementary materials. Our study included 81 adult SCD patients who are treated in different Hemoglobinopathies Units across Northern Greece. Their median age was 41 years, and 50 were female (61.7%). As expected in our population, the majority had the S/beta genotype (62), while 19 patients had the S/S genotype. Twenty-three presented SCD complications during the observation period (17 vaso-occlusive crises and six proteinuria/nephropathy) and were studied during this complication. Importantly, none of the patients that presented with renal damage, was on deferasirox, or other iron chelation therapy. The remaining 58 patients were studied at the end of the observation period. The comprehensive characteristics and parameters studied are summarized in Table 1.</p><p>First, we measured soluble C5b-9 and modified Ham test at a steady state and during the observation period. At steady state, soluble C5b-9 was above normal limits in a significantly higher percentage of patients who then presented a SCD complication compared to those who did not (7/23, 30%, <i>p</i> = 0.028). Similarly, patients who presented a complication had a significantly higher rate of modified Ham test (5/23, 21%, <i>p</i> = 0.001, Figure 1A) at steady state. During the observation period, we detected a significant increase in soluble C5b-9 (<i>p</i> < 0.001). This increase was significantly higher in patients with complications (<i>p</i> = 0.046, Figure 1B). One patient who did not experience complications had a positive Ham test and sC5b-9, while 42 patients who did not present complications had only elevated C5b-9. Since VOC was the most common complication, significant results were replicated in patients with VOC (data not shown).</p><p>ADAMTS13 was similar and within normal limits at steady state and follow-up (Figure 2B).</p><p>One other parameter studied as a marker of thromboinflammation was NETs. We were able to measure NETS on 60 of our 81 patients. NETs at steady state were significantly increased in patients who then developed complications related to the disease. Additionally, there was a significant increase of NETs measured at follow-up compared to steady state (<i>p</i> < 0.001, Figure 2B). Patients with a positive modified Ham test had increased C5b9 (<i>p</i> < 0.001). No significant association was found between NETs and C5b-9 in our population.</p><p>We started our genetic analysis from rare variants with a minor allele frequency (MAF) less than 1%, since rare variants have been commonly described in patients with complement-related disorders<span><sup>10</sup></span>. We detected 23 rare variants, as shown in detail in Supporting Information S1: Table 1. Almost all rare variants were documented in unique patients. Only rs35836460 in <i>CFH</i>, rs186530184 in <i>CFHR1</i>, rs183647515 in <i>THBD</i>, and rs202206149 in <i>ADAMTS13</i> were found in two patients, with rs3176136 in <i>THBD</i> detected in three patients. Among 23 rare variants, only three were characterized as pathogenic by bioinformatic tools as described in the next paragraph.</p><p>To further understand the clinical significance of the remaining detected variants, we used four bioinformatic tools. We found 8 missense variants characterized as deleterious by these tools, as presented in detail in Supporting Information S1: Table 2. Among them, three were also rare variants (rs117793540 in <i>C3</i>, rs1800579 in <i>THBD</i>, and rs143568784 in <i>ADAMTS13</i>), potentially confirming its pathogenic role. Each rare variant was found in one patient. Additionally, rs144082872 in <i>CFI</i> was also detected in one patient. The remaining deleterious variants were detected in 2–15 patients each. Patients were heterozygous, except for one homozygous for rs28647808 (<i>ADAMTS13</i>).</p><p>Next, we searched for detected variants in the database of complement gene variants. As shown in Supporting Information S1: Table 3, 21 variants have been described in this complement database. In agreement with the bioinformatic tools, rs144082872 in <i>CFI</i> has also been characterized as pathogenic in this database.</p><p>To better characterize the phenotype of SCD patients, we investigated associations of functional and genetic assays with clinical characteristics. All patients with pathogenic or deleterious variants had increased complement activation by functional assays (modified Ham test or soluble C5b9), except for two patients with CFB and two with ADAMTS13 variants. Interestingly, patients with a combination of pathogenic or deleterious variants had a significantly higher transfusion dependency rate (65% vs. 35%, <i>p</i> = 0.031).</p><p>Finally, we sought to determine predictors of complications. Among studied baseline patient characteristics (Supporting Information S1: Table 4), there was no univariate association with complications. Therefore, we included steady-state soluble C5b-9 and NETs, as well as the presence of a pathogenic or deleterious variant in multivariate analysis. The only independent predictor was C5b-9 levels.</p><p>In our study, we reveal activation of complement and thromboinflammation, even in the steady state of SCD patients who later develop complications. Notably, these markers experience a significant surge during the onset of complications. Germline genetic variants may contribute to the predisposition to complement activation, linked to clinical characteristics. Importantly, C5b-9 independently predicts complications in our multivariate analysis.</p><p>The inflammatory response becomes a key driver in SCD's pathophysiology, fueled by recurrent ischemic reperfusion events. Besides direct tissue damage, chronic inflammation contributes to cumulative damage in SCD. Complement activation plays a crucial role in both inflammation and the prominent hemolysis seen in SCD. Given the disease's multifactorial nature, markers such as NETs and ADAMTS13 are explored as potential treatment targets.</p><p>Assessing complement activation in clinical labs faces challenges due to limited access to robust assays.<span><sup>9</sup></span> Our study addresses this by introducing a modified Ham test for early and robust detection of complement activation, alongside confirming soluble C5b-9 as a practical marker for complications' evaluation. The increase in Bb fragments supports alternative pathway activation. Previous research using less precise assays indicated alternative pathway activation in SCD.<span><sup>11</sup></span></p><p>Exploring the genomic landscape of complement-related variants in SCD is a novel aspect of our study. Genetic analysis using multiple tools identifies pathogenic variants, highlighting an association between variants and clinical characteristics.<span><sup>12</sup></span> Our study has approached genetic analysis with multiple tools, while previous studies in complement-related disorders have reported only rare variants.<span><sup>13</sup></span> As our group has shown in thrombotic microangiopathies (TMA),<span><sup>14</sup></span> additional variants might also harbor clinical significance. In an effort to better understand the clinical significance of rare variants, novel databases have been created, such as the Database of Complement Gene Variants.<span><sup>12</sup></span> These databases are considerably helpful in linking the genotype with the phenotype in these patients. However, this approach does not take into account the majority of detected variants, whose functional and clinical significance remains to be studied. Variants such as rs1047286, rs2230199, rs800292 for C3, as well as rs1061147, rs1061170 for CFH, and the deleterious variant rs12614 for CFB have been implicated in the development of age-related macular degeneration (AMD).<span><sup>15, 16</sup></span></p><p>Current SCD pharmacological treatments, including hydroxyurea and voxelotor, show positive effects, but the need for novel therapeutic approaches persists.<span><sup>17</sup></span> Crizanlizumab, initially promising, was discontinued by the European Medicines Agency (EMA) due to efficacy issues.<span><sup>18</sup></span> Eculizumab, a complement inhibitor, shows encouraging results, but challenges hinder wider study.<span><sup>19</sup></span> Preclinical data suggest that MASP-2 or MASP-3 inhibition may prevent SCD complications.<span><sup>20</sup></span> The CROSSWALK trial is investigating the benefits of a C5 inhibitor in VOC prevention.<span><sup>21</sup></span></p><p>Our study has limitations, including varying patient treatments across centers and limited population size. Long-term observations in a larger cohort could provide additional insights. Data gaps on ADAMTS13Ag hinder a full understanding of its role in SCD. The predominantly S/β genotype in our population limits generalizability, and the impact of hydroxyurea on complement profiles is explored but inconclusive. The study's timing during the COVID-19 pandemic affected sample collection and patient visits. One other limitation is that 20 of our patients were on a chronic transfusion schedule. Moreover, our study's cohort is rather small, and further studies with bigger cohorts are needed to determine whether or not genetic variants play a role.</p><p>In conclusion, complement activation and thromboinflammation are evident in SCD patients, especially during complications. Considering the safety and efficacy of complement inhibitors in other complementopathies and preliminary data from SCD, our study highlights useful tools to early detect patients that might benefit from complement inhibition.</p><p>Christos Varelas and Eleni Gavriilaki designed research. Christos Varelas, Efthymia Vlachaki, Philippos Klonizakis, Despoina Pantelidou, Michael Diamantidis, Nikolaos Sabanis, Ioanna Christodoulou, Despina Papadopoulou, Evdoxia Koravou, Ioanna Sakellari, Stamatia Theodoridou, and Ioannis Mitroulis performed research. Christos Varelas, Fani Minti, Apostolia Papalexandri, Tasoula Touloumenidou, and Eleni Gavriilaki analyzed data and wrote the paper. Efthymia Vlachaki, Sofia Vakalopoulou, Vasilis Perifanis, George Vassilopoulos, and Eleni Gavriilaki contributed to the research design and edited and approved the paper.</p><p>Eleni Gavriilaki has consulted for Alexion, Omeros, and Sanofi Cooperation. The other authors declare no competing financial interest.</p>","PeriodicalId":12982,"journal":{"name":"HemaSphere","volume":"8 7","pages":""},"PeriodicalIF":7.6000,"publicationDate":"2024-07-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11270009/pdf/","citationCount":"0","resultStr":"{\"title\":\"Prospective study of complement activation and thromboinflammation within sickle cell disease and its complications\",\"authors\":\"Christos Varelas, Efthymia Vlachaki, Philippos Klonizakis, Despoina Pantelidou, Fani Minti, Michael Diamantidis, Nikolaos Sabanis, Evdoxia Koravou, Ioanna Christodoulou, Despina Papadopoulou, Stamatia Theodoridou, Tasoula Touloumenidou, Apostolia Papalexandri, Ioanna Sakellari, Sofia Vakalopoulou, Vasilis Perifanis, George Vassilopoulos, Ioannis Mitroulis, Eleni Gavriilaki\",\"doi\":\"10.1002/hem3.135\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Sickle cell disease (SCD) results from mutations in the β-globin gene, producing abnormal hemoglobin S (HbS) and leading to complications causing significant morbidity and mortality.<span><sup>1</sup></span> One of the hallmark consequences of SCD is the occurrence of vaso-occlusive crises (VOCs), which arise from the interplay of factors in the disease's pathophysiology, involving abnormal hemoglobin polymerization, inflammation, endothelial dysfunction, and activation of the immune system, culminating in the painful obstruction of blood vessels by sickled red blood cells, that tend to obstruct blood vessels, leading to reduced blood flow and oxygen supply. This vicious cycle of ischemia followed by reperfusion constitutes the ischemia-reperfusion model.<span><sup>2</sup></span></p><p>The complement system, a complex defense mechanism, is implicated in various diseases through unregulated activation.<span><sup>3</sup></span> However, diagnostic challenges hinder patient selection for complement inhibition.<span><sup>4</sup></span> Preliminary data from our group using novel assays indicate complement activation even at a steady state in a limited patient population.<span><sup>5</sup></span></p><p>Limited information exists on additional markers in the complement activation and endothelial dysfunction cycle in SCD. Neutrophil extracellular traps (NETs), indicative of thromboinflammation, are elevated in SCD patients, even during steady state.<span><sup>6</sup></span> ADAMTS13 (A Disintegrin and Metalloproteinase with Thrombospondin motifs), studied for its role in SCD vasculopathy, shows conflicting results as a potential biomarker.<span><sup>7, 8</sup></span> Genetic variants and autoantibodies leading to unregulated complement activation are implicated in the pathogenesis of various human diseases.<span><sup>9</sup></span></p><p>Despite the lack of specific biomarkers or targeted treatments for crises, our hypothesis posits the presence of complement activation and thromboinflammation in SCD, particularly during complications, with distinct yet unexplored clinical or genetic features in these patients.</p><p>Our study's methods regarding patient population, observation period, functional assays, and genetic, bioinformatic, and statistical analysis are demonstrated in supplementary materials. Our study included 81 adult SCD patients who are treated in different Hemoglobinopathies Units across Northern Greece. Their median age was 41 years, and 50 were female (61.7%). As expected in our population, the majority had the S/beta genotype (62), while 19 patients had the S/S genotype. Twenty-three presented SCD complications during the observation period (17 vaso-occlusive crises and six proteinuria/nephropathy) and were studied during this complication. Importantly, none of the patients that presented with renal damage, was on deferasirox, or other iron chelation therapy. The remaining 58 patients were studied at the end of the observation period. The comprehensive characteristics and parameters studied are summarized in Table 1.</p><p>First, we measured soluble C5b-9 and modified Ham test at a steady state and during the observation period. At steady state, soluble C5b-9 was above normal limits in a significantly higher percentage of patients who then presented a SCD complication compared to those who did not (7/23, 30%, <i>p</i> = 0.028). Similarly, patients who presented a complication had a significantly higher rate of modified Ham test (5/23, 21%, <i>p</i> = 0.001, Figure 1A) at steady state. During the observation period, we detected a significant increase in soluble C5b-9 (<i>p</i> < 0.001). This increase was significantly higher in patients with complications (<i>p</i> = 0.046, Figure 1B). One patient who did not experience complications had a positive Ham test and sC5b-9, while 42 patients who did not present complications had only elevated C5b-9. Since VOC was the most common complication, significant results were replicated in patients with VOC (data not shown).</p><p>ADAMTS13 was similar and within normal limits at steady state and follow-up (Figure 2B).</p><p>One other parameter studied as a marker of thromboinflammation was NETs. We were able to measure NETS on 60 of our 81 patients. NETs at steady state were significantly increased in patients who then developed complications related to the disease. Additionally, there was a significant increase of NETs measured at follow-up compared to steady state (<i>p</i> < 0.001, Figure 2B). Patients with a positive modified Ham test had increased C5b9 (<i>p</i> < 0.001). No significant association was found between NETs and C5b-9 in our population.</p><p>We started our genetic analysis from rare variants with a minor allele frequency (MAF) less than 1%, since rare variants have been commonly described in patients with complement-related disorders<span><sup>10</sup></span>. We detected 23 rare variants, as shown in detail in Supporting Information S1: Table 1. Almost all rare variants were documented in unique patients. Only rs35836460 in <i>CFH</i>, rs186530184 in <i>CFHR1</i>, rs183647515 in <i>THBD</i>, and rs202206149 in <i>ADAMTS13</i> were found in two patients, with rs3176136 in <i>THBD</i> detected in three patients. Among 23 rare variants, only three were characterized as pathogenic by bioinformatic tools as described in the next paragraph.</p><p>To further understand the clinical significance of the remaining detected variants, we used four bioinformatic tools. We found 8 missense variants characterized as deleterious by these tools, as presented in detail in Supporting Information S1: Table 2. Among them, three were also rare variants (rs117793540 in <i>C3</i>, rs1800579 in <i>THBD</i>, and rs143568784 in <i>ADAMTS13</i>), potentially confirming its pathogenic role. Each rare variant was found in one patient. Additionally, rs144082872 in <i>CFI</i> was also detected in one patient. The remaining deleterious variants were detected in 2–15 patients each. Patients were heterozygous, except for one homozygous for rs28647808 (<i>ADAMTS13</i>).</p><p>Next, we searched for detected variants in the database of complement gene variants. As shown in Supporting Information S1: Table 3, 21 variants have been described in this complement database. In agreement with the bioinformatic tools, rs144082872 in <i>CFI</i> has also been characterized as pathogenic in this database.</p><p>To better characterize the phenotype of SCD patients, we investigated associations of functional and genetic assays with clinical characteristics. All patients with pathogenic or deleterious variants had increased complement activation by functional assays (modified Ham test or soluble C5b9), except for two patients with CFB and two with ADAMTS13 variants. Interestingly, patients with a combination of pathogenic or deleterious variants had a significantly higher transfusion dependency rate (65% vs. 35%, <i>p</i> = 0.031).</p><p>Finally, we sought to determine predictors of complications. Among studied baseline patient characteristics (Supporting Information S1: Table 4), there was no univariate association with complications. Therefore, we included steady-state soluble C5b-9 and NETs, as well as the presence of a pathogenic or deleterious variant in multivariate analysis. The only independent predictor was C5b-9 levels.</p><p>In our study, we reveal activation of complement and thromboinflammation, even in the steady state of SCD patients who later develop complications. Notably, these markers experience a significant surge during the onset of complications. Germline genetic variants may contribute to the predisposition to complement activation, linked to clinical characteristics. Importantly, C5b-9 independently predicts complications in our multivariate analysis.</p><p>The inflammatory response becomes a key driver in SCD's pathophysiology, fueled by recurrent ischemic reperfusion events. Besides direct tissue damage, chronic inflammation contributes to cumulative damage in SCD. Complement activation plays a crucial role in both inflammation and the prominent hemolysis seen in SCD. Given the disease's multifactorial nature, markers such as NETs and ADAMTS13 are explored as potential treatment targets.</p><p>Assessing complement activation in clinical labs faces challenges due to limited access to robust assays.<span><sup>9</sup></span> Our study addresses this by introducing a modified Ham test for early and robust detection of complement activation, alongside confirming soluble C5b-9 as a practical marker for complications' evaluation. The increase in Bb fragments supports alternative pathway activation. Previous research using less precise assays indicated alternative pathway activation in SCD.<span><sup>11</sup></span></p><p>Exploring the genomic landscape of complement-related variants in SCD is a novel aspect of our study. Genetic analysis using multiple tools identifies pathogenic variants, highlighting an association between variants and clinical characteristics.<span><sup>12</sup></span> Our study has approached genetic analysis with multiple tools, while previous studies in complement-related disorders have reported only rare variants.<span><sup>13</sup></span> As our group has shown in thrombotic microangiopathies (TMA),<span><sup>14</sup></span> additional variants might also harbor clinical significance. In an effort to better understand the clinical significance of rare variants, novel databases have been created, such as the Database of Complement Gene Variants.<span><sup>12</sup></span> These databases are considerably helpful in linking the genotype with the phenotype in these patients. However, this approach does not take into account the majority of detected variants, whose functional and clinical significance remains to be studied. Variants such as rs1047286, rs2230199, rs800292 for C3, as well as rs1061147, rs1061170 for CFH, and the deleterious variant rs12614 for CFB have been implicated in the development of age-related macular degeneration (AMD).<span><sup>15, 16</sup></span></p><p>Current SCD pharmacological treatments, including hydroxyurea and voxelotor, show positive effects, but the need for novel therapeutic approaches persists.<span><sup>17</sup></span> Crizanlizumab, initially promising, was discontinued by the European Medicines Agency (EMA) due to efficacy issues.<span><sup>18</sup></span> Eculizumab, a complement inhibitor, shows encouraging results, but challenges hinder wider study.<span><sup>19</sup></span> Preclinical data suggest that MASP-2 or MASP-3 inhibition may prevent SCD complications.<span><sup>20</sup></span> The CROSSWALK trial is investigating the benefits of a C5 inhibitor in VOC prevention.<span><sup>21</sup></span></p><p>Our study has limitations, including varying patient treatments across centers and limited population size. Long-term observations in a larger cohort could provide additional insights. Data gaps on ADAMTS13Ag hinder a full understanding of its role in SCD. The predominantly S/β genotype in our population limits generalizability, and the impact of hydroxyurea on complement profiles is explored but inconclusive. The study's timing during the COVID-19 pandemic affected sample collection and patient visits. One other limitation is that 20 of our patients were on a chronic transfusion schedule. Moreover, our study's cohort is rather small, and further studies with bigger cohorts are needed to determine whether or not genetic variants play a role.</p><p>In conclusion, complement activation and thromboinflammation are evident in SCD patients, especially during complications. Considering the safety and efficacy of complement inhibitors in other complementopathies and preliminary data from SCD, our study highlights useful tools to early detect patients that might benefit from complement inhibition.</p><p>Christos Varelas and Eleni Gavriilaki designed research. Christos Varelas, Efthymia Vlachaki, Philippos Klonizakis, Despoina Pantelidou, Michael Diamantidis, Nikolaos Sabanis, Ioanna Christodoulou, Despina Papadopoulou, Evdoxia Koravou, Ioanna Sakellari, Stamatia Theodoridou, and Ioannis Mitroulis performed research. Christos Varelas, Fani Minti, Apostolia Papalexandri, Tasoula Touloumenidou, and Eleni Gavriilaki analyzed data and wrote the paper. Efthymia Vlachaki, Sofia Vakalopoulou, Vasilis Perifanis, George Vassilopoulos, and Eleni Gavriilaki contributed to the research design and edited and approved the paper.</p><p>Eleni Gavriilaki has consulted for Alexion, Omeros, and Sanofi Cooperation. The other authors declare no competing financial interest.</p>\",\"PeriodicalId\":12982,\"journal\":{\"name\":\"HemaSphere\",\"volume\":\"8 7\",\"pages\":\"\"},\"PeriodicalIF\":7.6000,\"publicationDate\":\"2024-07-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11270009/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"HemaSphere\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/hem3.135\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"HemaSphere","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/hem3.135","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
Prospective study of complement activation and thromboinflammation within sickle cell disease and its complications
Sickle cell disease (SCD) results from mutations in the β-globin gene, producing abnormal hemoglobin S (HbS) and leading to complications causing significant morbidity and mortality.1 One of the hallmark consequences of SCD is the occurrence of vaso-occlusive crises (VOCs), which arise from the interplay of factors in the disease's pathophysiology, involving abnormal hemoglobin polymerization, inflammation, endothelial dysfunction, and activation of the immune system, culminating in the painful obstruction of blood vessels by sickled red blood cells, that tend to obstruct blood vessels, leading to reduced blood flow and oxygen supply. This vicious cycle of ischemia followed by reperfusion constitutes the ischemia-reperfusion model.2
The complement system, a complex defense mechanism, is implicated in various diseases through unregulated activation.3 However, diagnostic challenges hinder patient selection for complement inhibition.4 Preliminary data from our group using novel assays indicate complement activation even at a steady state in a limited patient population.5
Limited information exists on additional markers in the complement activation and endothelial dysfunction cycle in SCD. Neutrophil extracellular traps (NETs), indicative of thromboinflammation, are elevated in SCD patients, even during steady state.6 ADAMTS13 (A Disintegrin and Metalloproteinase with Thrombospondin motifs), studied for its role in SCD vasculopathy, shows conflicting results as a potential biomarker.7, 8 Genetic variants and autoantibodies leading to unregulated complement activation are implicated in the pathogenesis of various human diseases.9
Despite the lack of specific biomarkers or targeted treatments for crises, our hypothesis posits the presence of complement activation and thromboinflammation in SCD, particularly during complications, with distinct yet unexplored clinical or genetic features in these patients.
Our study's methods regarding patient population, observation period, functional assays, and genetic, bioinformatic, and statistical analysis are demonstrated in supplementary materials. Our study included 81 adult SCD patients who are treated in different Hemoglobinopathies Units across Northern Greece. Their median age was 41 years, and 50 were female (61.7%). As expected in our population, the majority had the S/beta genotype (62), while 19 patients had the S/S genotype. Twenty-three presented SCD complications during the observation period (17 vaso-occlusive crises and six proteinuria/nephropathy) and were studied during this complication. Importantly, none of the patients that presented with renal damage, was on deferasirox, or other iron chelation therapy. The remaining 58 patients were studied at the end of the observation period. The comprehensive characteristics and parameters studied are summarized in Table 1.
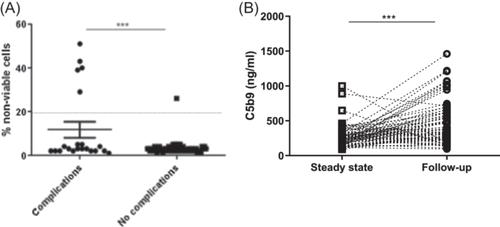
First, we measured soluble C5b-9 and modified Ham test at a steady state and during the observation period. At steady state, soluble C5b-9 was above normal limits in a significantly higher percentage of patients who then presented a SCD complication compared to those who did not (7/23, 30%, p = 0.028). Similarly, patients who presented a complication had a significantly higher rate of modified Ham test (5/23, 21%, p = 0.001, Figure 1A) at steady state. During the observation period, we detected a significant increase in soluble C5b-9 (p < 0.001). This increase was significantly higher in patients with complications (p = 0.046, Figure 1B). One patient who did not experience complications had a positive Ham test and sC5b-9, while 42 patients who did not present complications had only elevated C5b-9. Since VOC was the most common complication, significant results were replicated in patients with VOC (data not shown).
ADAMTS13 was similar and within normal limits at steady state and follow-up (Figure 2B).
One other parameter studied as a marker of thromboinflammation was NETs. We were able to measure NETS on 60 of our 81 patients. NETs at steady state were significantly increased in patients who then developed complications related to the disease. Additionally, there was a significant increase of NETs measured at follow-up compared to steady state (p < 0.001, Figure 2B). Patients with a positive modified Ham test had increased C5b9 (p < 0.001). No significant association was found between NETs and C5b-9 in our population.
We started our genetic analysis from rare variants with a minor allele frequency (MAF) less than 1%, since rare variants have been commonly described in patients with complement-related disorders10. We detected 23 rare variants, as shown in detail in Supporting Information S1: Table 1. Almost all rare variants were documented in unique patients. Only rs35836460 in CFH, rs186530184 in CFHR1, rs183647515 in THBD, and rs202206149 in ADAMTS13 were found in two patients, with rs3176136 in THBD detected in three patients. Among 23 rare variants, only three were characterized as pathogenic by bioinformatic tools as described in the next paragraph.
To further understand the clinical significance of the remaining detected variants, we used four bioinformatic tools. We found 8 missense variants characterized as deleterious by these tools, as presented in detail in Supporting Information S1: Table 2. Among them, three were also rare variants (rs117793540 in C3, rs1800579 in THBD, and rs143568784 in ADAMTS13), potentially confirming its pathogenic role. Each rare variant was found in one patient. Additionally, rs144082872 in CFI was also detected in one patient. The remaining deleterious variants were detected in 2–15 patients each. Patients were heterozygous, except for one homozygous for rs28647808 (ADAMTS13).
Next, we searched for detected variants in the database of complement gene variants. As shown in Supporting Information S1: Table 3, 21 variants have been described in this complement database. In agreement with the bioinformatic tools, rs144082872 in CFI has also been characterized as pathogenic in this database.
To better characterize the phenotype of SCD patients, we investigated associations of functional and genetic assays with clinical characteristics. All patients with pathogenic or deleterious variants had increased complement activation by functional assays (modified Ham test or soluble C5b9), except for two patients with CFB and two with ADAMTS13 variants. Interestingly, patients with a combination of pathogenic or deleterious variants had a significantly higher transfusion dependency rate (65% vs. 35%, p = 0.031).
Finally, we sought to determine predictors of complications. Among studied baseline patient characteristics (Supporting Information S1: Table 4), there was no univariate association with complications. Therefore, we included steady-state soluble C5b-9 and NETs, as well as the presence of a pathogenic or deleterious variant in multivariate analysis. The only independent predictor was C5b-9 levels.
In our study, we reveal activation of complement and thromboinflammation, even in the steady state of SCD patients who later develop complications. Notably, these markers experience a significant surge during the onset of complications. Germline genetic variants may contribute to the predisposition to complement activation, linked to clinical characteristics. Importantly, C5b-9 independently predicts complications in our multivariate analysis.
The inflammatory response becomes a key driver in SCD's pathophysiology, fueled by recurrent ischemic reperfusion events. Besides direct tissue damage, chronic inflammation contributes to cumulative damage in SCD. Complement activation plays a crucial role in both inflammation and the prominent hemolysis seen in SCD. Given the disease's multifactorial nature, markers such as NETs and ADAMTS13 are explored as potential treatment targets.
Assessing complement activation in clinical labs faces challenges due to limited access to robust assays.9 Our study addresses this by introducing a modified Ham test for early and robust detection of complement activation, alongside confirming soluble C5b-9 as a practical marker for complications' evaluation. The increase in Bb fragments supports alternative pathway activation. Previous research using less precise assays indicated alternative pathway activation in SCD.11
Exploring the genomic landscape of complement-related variants in SCD is a novel aspect of our study. Genetic analysis using multiple tools identifies pathogenic variants, highlighting an association between variants and clinical characteristics.12 Our study has approached genetic analysis with multiple tools, while previous studies in complement-related disorders have reported only rare variants.13 As our group has shown in thrombotic microangiopathies (TMA),14 additional variants might also harbor clinical significance. In an effort to better understand the clinical significance of rare variants, novel databases have been created, such as the Database of Complement Gene Variants.12 These databases are considerably helpful in linking the genotype with the phenotype in these patients. However, this approach does not take into account the majority of detected variants, whose functional and clinical significance remains to be studied. Variants such as rs1047286, rs2230199, rs800292 for C3, as well as rs1061147, rs1061170 for CFH, and the deleterious variant rs12614 for CFB have been implicated in the development of age-related macular degeneration (AMD).15, 16
Current SCD pharmacological treatments, including hydroxyurea and voxelotor, show positive effects, but the need for novel therapeutic approaches persists.17 Crizanlizumab, initially promising, was discontinued by the European Medicines Agency (EMA) due to efficacy issues.18 Eculizumab, a complement inhibitor, shows encouraging results, but challenges hinder wider study.19 Preclinical data suggest that MASP-2 or MASP-3 inhibition may prevent SCD complications.20 The CROSSWALK trial is investigating the benefits of a C5 inhibitor in VOC prevention.21
Our study has limitations, including varying patient treatments across centers and limited population size. Long-term observations in a larger cohort could provide additional insights. Data gaps on ADAMTS13Ag hinder a full understanding of its role in SCD. The predominantly S/β genotype in our population limits generalizability, and the impact of hydroxyurea on complement profiles is explored but inconclusive. The study's timing during the COVID-19 pandemic affected sample collection and patient visits. One other limitation is that 20 of our patients were on a chronic transfusion schedule. Moreover, our study's cohort is rather small, and further studies with bigger cohorts are needed to determine whether or not genetic variants play a role.
In conclusion, complement activation and thromboinflammation are evident in SCD patients, especially during complications. Considering the safety and efficacy of complement inhibitors in other complementopathies and preliminary data from SCD, our study highlights useful tools to early detect patients that might benefit from complement inhibition.
Christos Varelas and Eleni Gavriilaki designed research. Christos Varelas, Efthymia Vlachaki, Philippos Klonizakis, Despoina Pantelidou, Michael Diamantidis, Nikolaos Sabanis, Ioanna Christodoulou, Despina Papadopoulou, Evdoxia Koravou, Ioanna Sakellari, Stamatia Theodoridou, and Ioannis Mitroulis performed research. Christos Varelas, Fani Minti, Apostolia Papalexandri, Tasoula Touloumenidou, and Eleni Gavriilaki analyzed data and wrote the paper. Efthymia Vlachaki, Sofia Vakalopoulou, Vasilis Perifanis, George Vassilopoulos, and Eleni Gavriilaki contributed to the research design and edited and approved the paper.
Eleni Gavriilaki has consulted for Alexion, Omeros, and Sanofi Cooperation. The other authors declare no competing financial interest.
期刊介绍:
HemaSphere, as a publication, is dedicated to disseminating the outcomes of profoundly pertinent basic, translational, and clinical research endeavors within the field of hematology. The journal actively seeks robust studies that unveil novel discoveries with significant ramifications for hematology.
In addition to original research, HemaSphere features review articles and guideline articles that furnish lucid synopses and discussions of emerging developments, along with recommendations for patient care.
Positioned as the foremost resource in hematology, HemaSphere augments its offerings with specialized sections like HemaTopics and HemaPolicy. These segments engender insightful dialogues covering a spectrum of hematology-related topics, including digestible summaries of pivotal articles, updates on new therapies, deliberations on European policy matters, and other noteworthy news items within the field. Steering the course of HemaSphere are Editor in Chief Jan Cools and Deputy Editor in Chief Claire Harrison, alongside the guidance of an esteemed Editorial Board comprising international luminaries in both research and clinical realms, each representing diverse areas of hematologic expertise.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


